



Vol 69, No 2 (2024)

- Year: 2024
- Published: 06.05.2024
- Articles: 9
- URL: https://virusjour.crie.ru/jour/issue/view/133
Full Issue

EDITORIAL CONCEPT
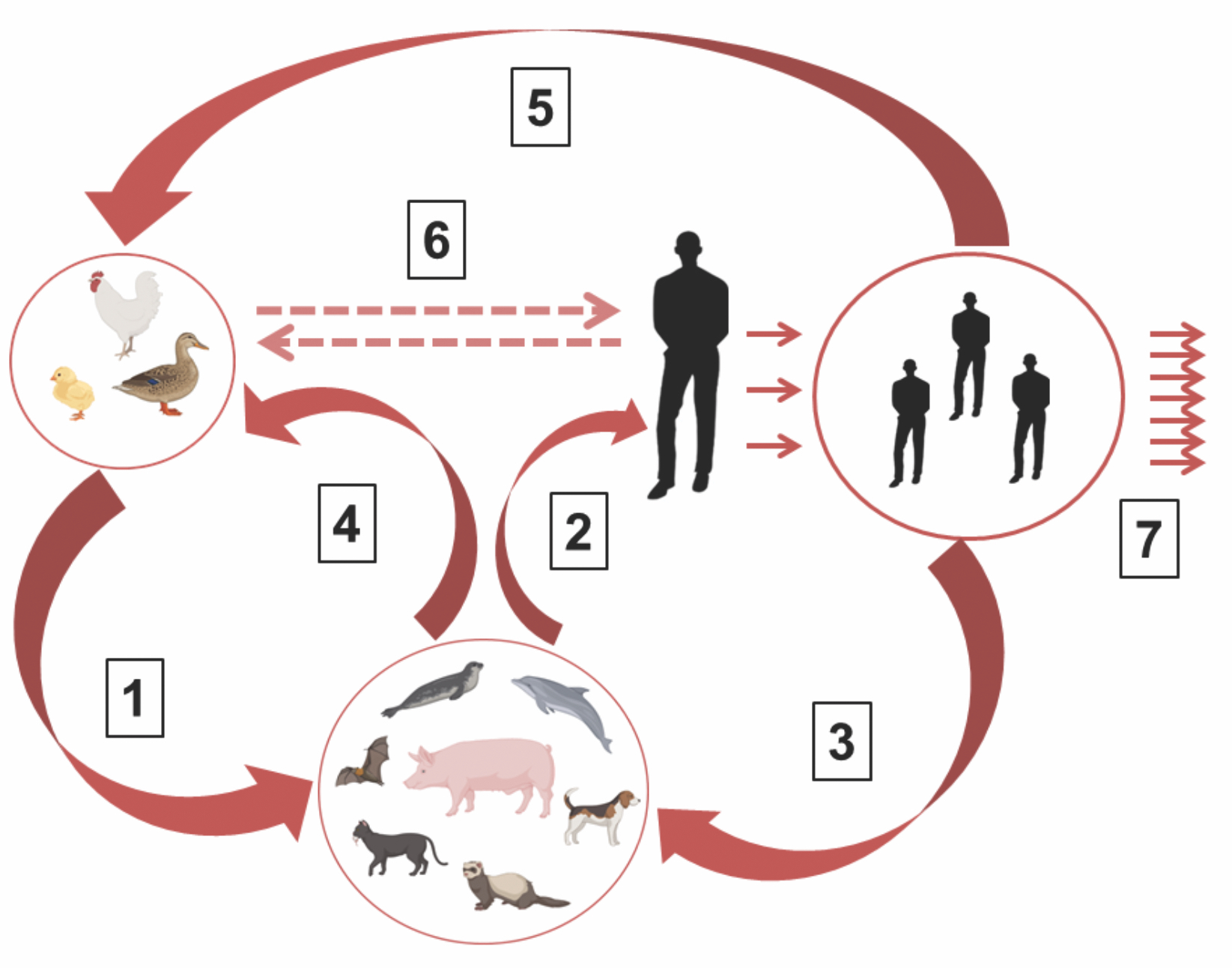
Avian flu: «for whom the bell tolls»?
Abstract
The family Orthomyxoviridae consists of 9 genera, including Alphainfluenza virus, which contains avian influenza viruses. In two subtypes H5 and H7 besides common low-virulent strains, a specific type of highly virulent avian virus have been described to cause more than 60% mortality among domestic birds. These variants of influenza virus are usually referred to as «avian influenza virus». The difference between high (HPAI) and low (LPAI) virulent influenza viruses is due to the structure of the arginine-containing proteolytic activation site in the hemagglutinin (HA) protein. The highly virulent avian influenza virus H5 was identified more than 100 years ago and during this time they cause outbreaks among wild and domestic birds on all continents and only a few local episodes of the disease in humans have been identified in XXI century. Currently, a sharp increase in the incidence of highly virulent virus of the H5N1 subtype (clade h2.3.4.4b) has been registered in birds on all continents, accompanied by the transmission of the virus to various species of mammals. The recorded global mortality rate among wild, domestic and agricultural birds from H5 subtype is approaching to the level of 1 billion cases. A dangerous epidemic factor is becoming more frequent outbreaks of avian influenza with high mortality among mammals, in particular seals and marine lions in North and South America, minks and fur-bearing animals in Spain and Finland, domestic and street cats in Poland. H5N1 avian influenza clade h2.3.4.4b strains isolated from mammals have genetic signatures of partial adaptation to the human body in the PB2, NP, HA, NA genes, which play a major role in regulating the aerosol transmission and the host range of the virus. The current situation poses a real threat of pre-adaptation of the virus in mammals as intermediate hosts, followed by the transition of the pre-adapted virus into the human population with catastrophic consequences.
 101-118
101-118



REVIEWS
The use of immunoglobulins and monoclonal antibodies against COVID-19
Abstract
Introduction. When a new disease occurs, one of the most affordable remedies is drugs containing specific antibodies to this infectious agent. The use of such drugs is aimed at reducing the amount of the pathogen in the macroorganism and the associated reduction in the severity of the symptoms of the disease or recovery.
The purpose of this review is to analyze the experience of using immunoglobulins and monoclonal antibodies in the treatment of COVID-19 patients during the pandemic.
Results and conclusion. The two main groups of medical protective agents that block the penetration of the SARS-CoV-2 virus into permissive cells are drugs obtained from blood plasma of convalescents (immunoglobulin) and human monoclonal antibodies. The first group of drugs in the treatment of COVID-19 includes blood plasma of convalescents, which can be successfully used for emergency prevention. The main disadvantage of using blood plasma convalescents is the difficulty of standardization due to the different content of specific antibodies in donors. Another disadvantage is the undesirable side effects in recipients that occur after plasma administration. An alternative approach to COVID-19 therapy is the use of humanized and genetically engineered human monoclonal antibodies against certain epitopes of the SARS-CoV-2 virus. For example, monoclonal antibodies against receptor-binding domain of the S-protein, which prevents the virus from entering permissive cells and interrupts the development of infection. The advantages of these drugs are their safety, high specific activity, and the possibility of standardization. However, the complexity of their production and high cost make them inaccessible for mass use in practical medicine.
119-126
ORIGINAL RESEARCHES
Gene expression profiling of p53 and c-myc in HTLV-1 positive blood donors in Congo
Abstract
Objectives. The HTLV-1 infection persists for life, remaining as asymptomatic viral reservoirs in most patients, ensuring the chain of transmission, but around 4% develop adult T-cell leukemia/lymphoma (ATLL). HTLV-1 is an oncogenic retrovirus that transforms CD4+ T lymphocytes and deregulates the lymphoproliferative pathways that contribute to the development of ATLL. To achieve cell transformation, most oncogenic retroviruses use proto-oncogene capture transduction, with proviral integration disrupting the expression of tumor suppressors or proto-oncogenes.
The aim. We conducted this study on the prevalence of HTLV-1 infection in blood donors to expand the HTLV-1 database, assess the risk of transmission via blood products, as well as evaluate the risk of persistent infection or development of neoplastic diseases in HTLV-1 carriers.
Materials and methods. This is a cross-sectional study of blood donors of all categories. For this study, 265 blood donors were recruited at the Centre National de Transfusion Sanguine in Brazzaville. After testing for HTLV-1 antibodies by ELISA, proviral DNA was extracted from all ELISA-positive samples for detection by nested PCR, followed by RT qPCR using specific primers p53 and c-myc for gene expression.
Results. 20/265 were positive for anti-HTLV-1 antibody, 5 donors were positive for proviral DNA. The prevalence of HTLV-1 was 1.8%. All HTLV-1-positive donors were male (1.8%), with a positive correlation (p = 0.05); the 1.1% of positive donors were regular, with the majority aged between 31 and 45 years (1.5%), and concubine donors were the most frequent (1.1%). All samples showed normal expression of the p53 and c-myc genes.
Conclusion. The prevalence, though low, remains a serious problem. No abnormal p53 or c-myc gene expression was detected in HTLV-1-positive donors, which could mean that none of the T lymphocytes in these donors had been transformed by HTLV-1.
127-133
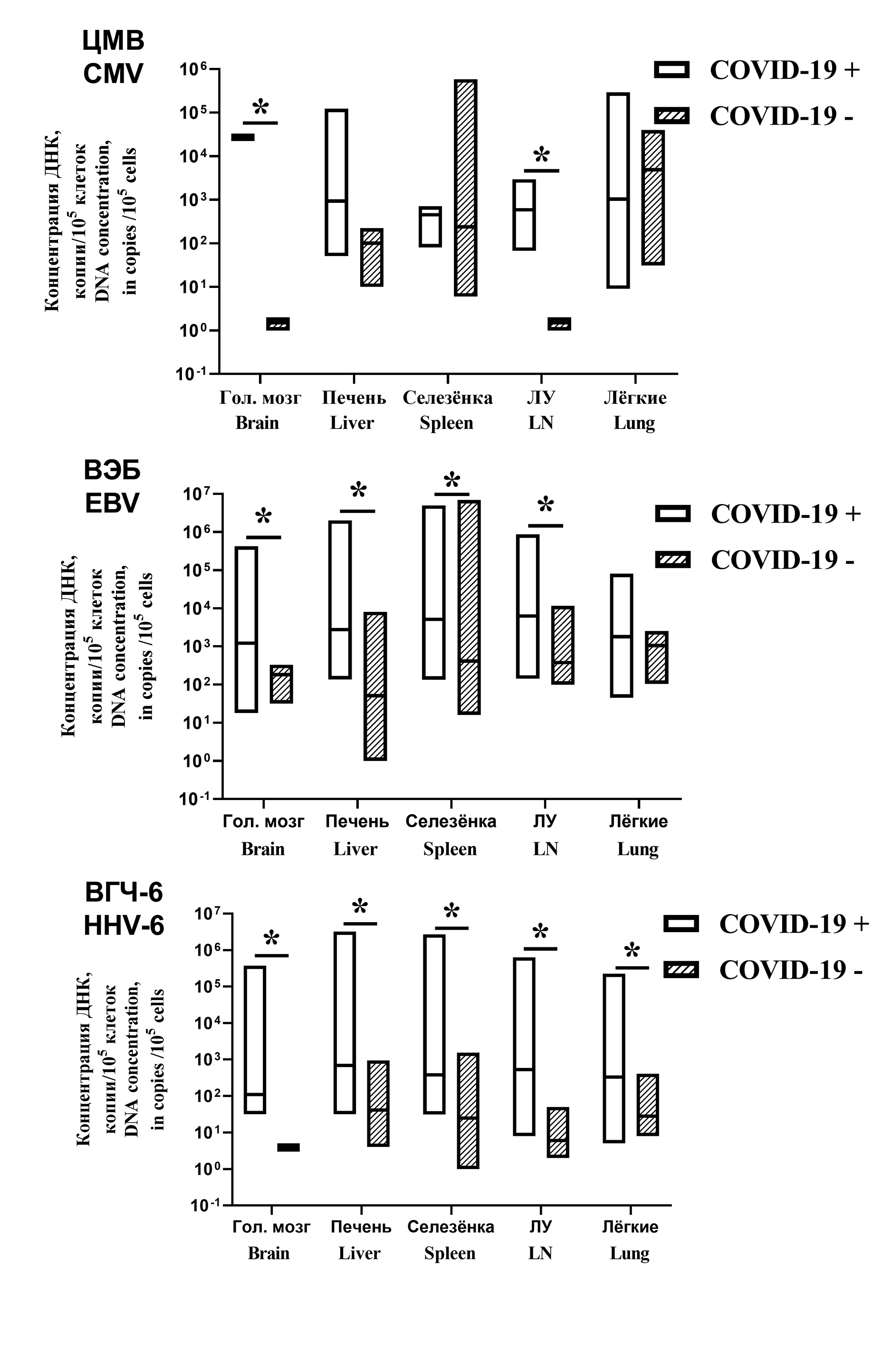
Detection rates and high concentration of herpesvirus (Orthoherpesviridae) DNA in autopsy materials from patients with COVID-19 fatal outcome
Abstract
Introduction. SARS-CoV-2 infection causes immune disorders that create conditions for the reactivation of human herpesviruses (HHVs). However, the estimates of the HHVs effect on the course and outcome of COVID-19 are ambiguous.
Аim – to study the possible relationship between the HHV reactivation and the adverse outcome of COVID-19.
Materials and methods. Postmortem samples from the brain, liver, spleen, lymph nodes and lungs were obtained from 59 patients treated at the Moscow Infectious Diseases Hospital No.1 in 2021–2023. The group 1 comprised 39 patients with fatal COVID-19; group 2 (comparison group) included 20 patients not infected with SARS-CoV-2 who died from various somatic diseases. HHV DNA and SARS-CoV-2 RNA were determined by PCR.
Results. HHV DNA was found in autopsy samples from all patients. In group 1, EBV was most often detected in lymph nodes (94%), HHV-6 in liver (68%), CMV in lymph nodes (18%), HSV in brain (16%), VZV in lung and spleen (3% each). The detection rates of HHVs in both groups was similar. Important differences were found in viral load. In patients with COVID-19, the number of samples containing more than 1,000 copies of HHV DNA per 100,000 cells was 52.4%, in the comparison group – 16.6% (p < 0.002). An association has been established between the reactivation of HSV and HHV-6 and the severity of lung damage. Reactivation of EBV correlated with increased levels of liver enzymes.
Conclusion. Reactivation of HHVs in patients with fatal COVID-19 was associated with severe lung and liver damages, which indicates a link between HHV reactivation and COVID-19 deaths.
134-150
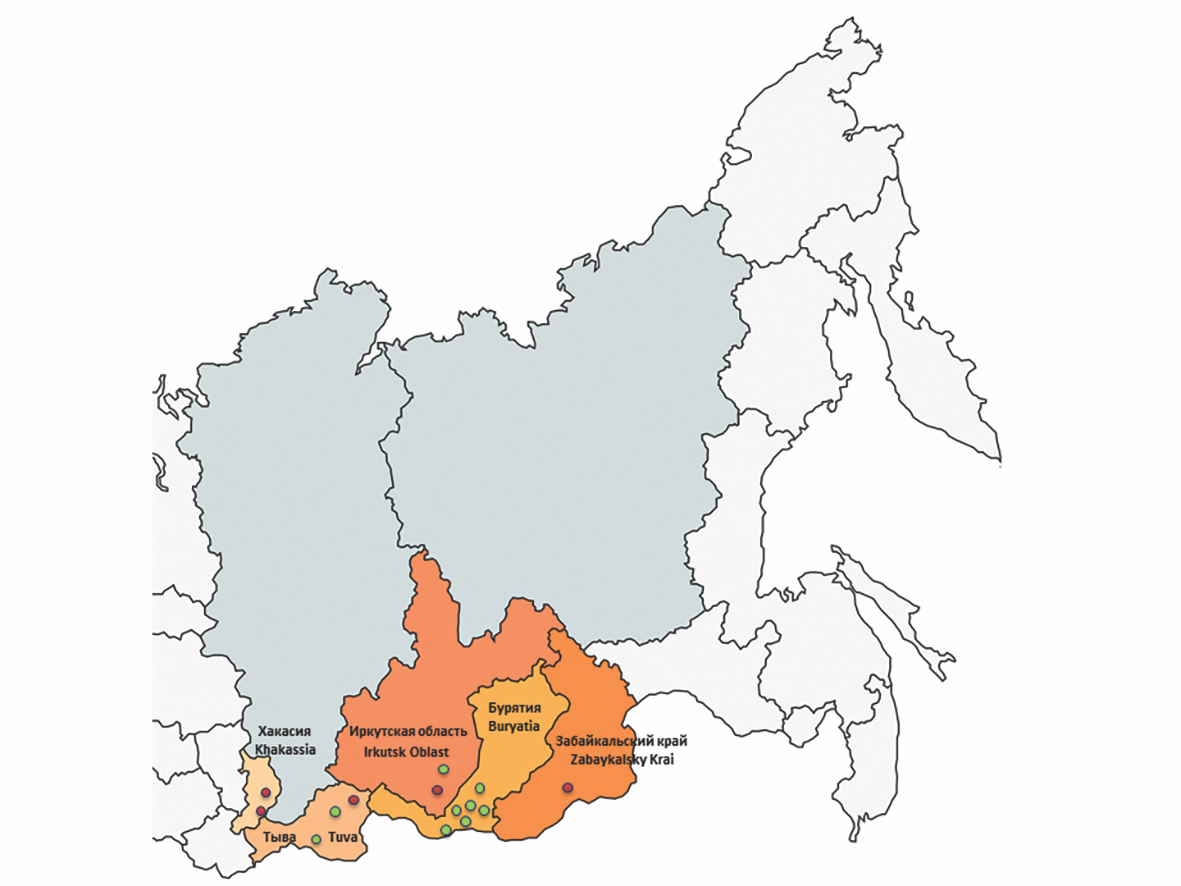
Prevalence and genetic diversity of the Alongshan virus (Flaviviridae) circulating in ticks in the south of Eastern Siberia
Abstract
Introduction. Tick-borne infections are of great importance for many regions of Russia, including Eastern Siberia. This unfavorable epidemiological situation can be characterized not only by the circulation of well-known tick-borne infections, but also by the identification of new pathogens, the role of which remains little or generally unexplored. Multicomponent flavi-like viruses can cause infectious diseases in humans and pose a threat to public health.
The purpose of the study was the identification and molecular genetic characterization of the Alongshan virus (Flaviviridae, ALSV) isolates, transmitted by ticks in the south of Eastern Siberia.
Materials and methods. Total 1060 ticks were collected and analyzed from the territory of the Republics of Khakassia, Tuva, Buryatia, Irkutsk Region and Transbaikal Territory (Zabaykalsky Krai) in the spring-summer period 2023. ALSV RNA was detected by RT-PCR followed by nucleotide sequence determination and phylogenetic analysis for each segment of the genome.
Results. The ALSV infection rate in Ixodes persulcatus ticks collected in the Republic of Khakassia was 3.3% (95% CI: 1.4–7.5); in Irkutsk Oblast – 1.0% (95% CI: 0.3–3.7); in the Republic of Tuva – 0.9% (95% CI: 0.3–3.4) and in Transbaikal Krai – 0.7% (95% CI: 0.2–3.6). Sequences of all four segments of ALSV genetic variants circulating in I. persulcatus ticks in the south of Eastern Siberia are grouped with sequences found in China and clustered into the Asian subgroup transmitted by taiga ticks. The level of difference in the nucleotide sequences of genome fragments among the identified genetic variants of ALSV ranged from 2 to 3%.
Conclusion. The article shows the widespread distribution of ALSV in I. persulcatus ticks in the Republics of Khakassia and Tyva, Irkutsk Oblast and Transbaikal Territory. The obtained data actualize monitoring of changes in the area of distribution of potentially dangerous for humans flavi-like viruses and their vectors.
151-161
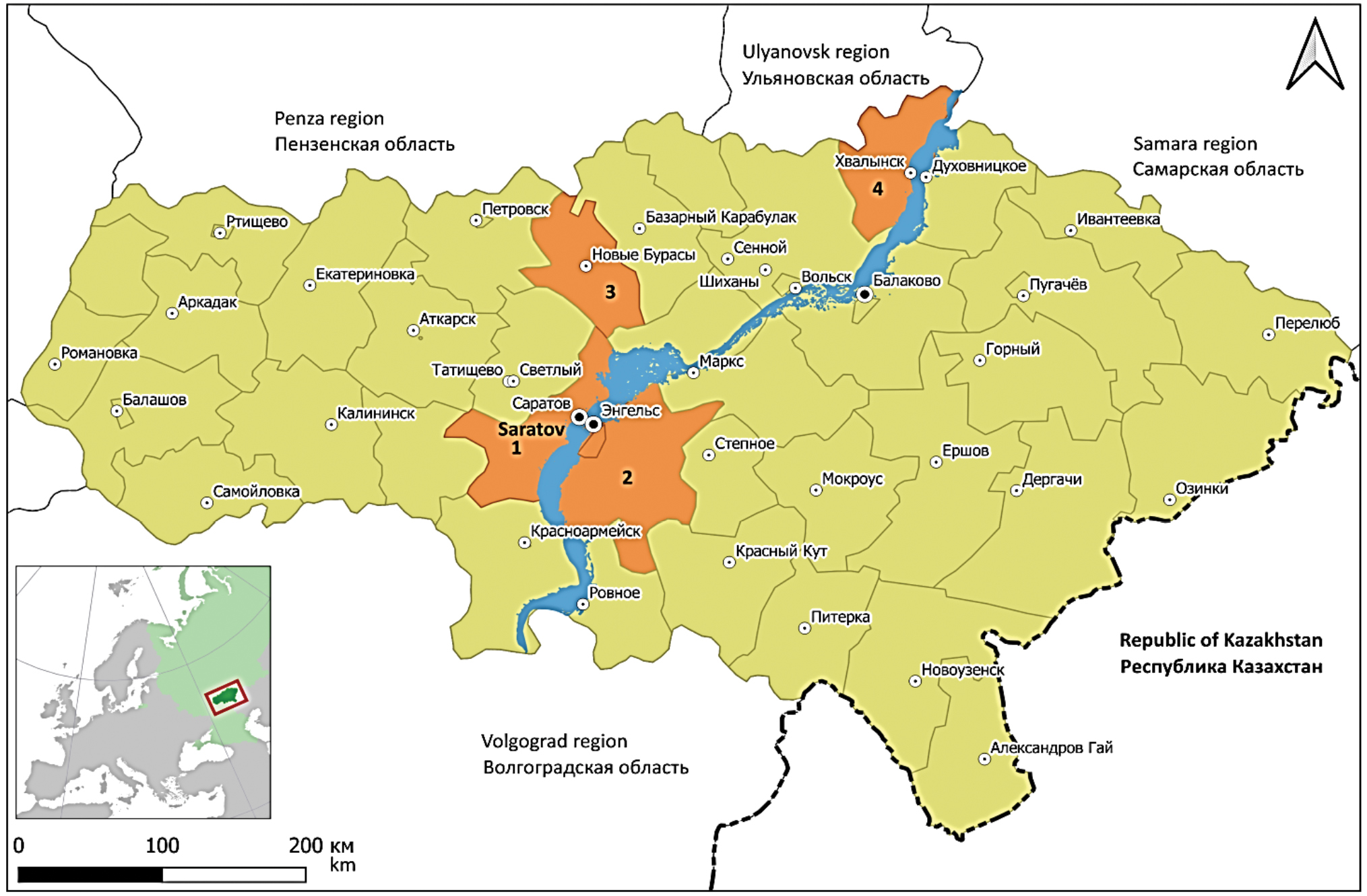
Phylogenetic analysis of variants of the Puumala virus (Hantaviridae: Orthohantavirus) circulating in the Saratov region
Abstract
The objective is to determine the complete nucleotide sequence and conduct a phylogenetic analysis of genome variants of the Puumala virus isolated in the Saratov region.
Materials and methods. The samples for the study were field material collected in the Gagarinsky (formerly Saratovsky), Engelssky, Novoburassky and Khvalynsky districts of the Saratov region in the period from 2019 to 2022. To specifically enrich the Puumala virus genome in the samples, were used PCR and developed a specific primer panel. Next, the resulting PCR products were sequenced and the fragments were assembled into one sequence for each segment of the virus genome. To construct phylogenetic trees, the maximum parsimony algorithm was used.
Results. Genetic variants of the Puumala virus isolated in the Saratov region have a high degree of genome similarity to each other, which indicates their unity of origin. According to phylogenetic analysis, they all form a separate branch in the cluster formed by hantaviruses from other subjects of the Volga Federal District. The virus variants from the Republics of Udmurtia and Tatarstan, as well as from the Samara and Ulyanovsk regions, are closest to the samples from the Saratov region.
Conclusion. The data obtained show the presence of a pronounced territorial confinement of strains to certain regions or areas that are the natural biotopes of their carriers. This makes it possible to fairly accurately determine the territory of possible infection of patients and/or the circulation of carriers of these virus variants based on the sequence of individual segments of their genome.
162-174
Development, production and characterization of SARS-CoV-2 virus-like particles (Coronaviridae: Orthocoronavirinae: Betacoronavirus: Sarbecovirus)
Abstract
Introduction. The COVID-19 pandemic caused by SARS-CoV-2 has created serious health problems worldwide. The most effective way to prevent the occurrence of new epidemic outbreaks is vaccination. One of the modern and effective approaches to vaccine development is the use of virus-like particles (VLPs).
The aim of the study is to develop a technology for production of VLP based on recombinant SARS-CoV-2 proteins (E, M, N and S) in insect cells.
Materials and methods. Synthetic genes encoding coronavirus proteins E, M, N and S were used. VLP with various surface proteins of strains similar to the Wuhan virus, Delta, Alpha and Omicron were developed and cloned into the pFastBac plasmid. The proteins were synthesized in the baculovirus expression system and assembled into VLP in the portable Trichoplusia ni cell. The presence of insertion in the baculovirus genome was determined by PCR. ELISA and immunoblotting were used to study the antigenic activity of VLP. VLP purification was performed by ultracentrifugation using 20% sucrose. Morphology was assessed using electron microscopy and dynamic light scattering.
Results. VLPs consisting of recombinant SARS-CoV-2 proteins (S, M, E and N) were obtained and characterized. The specific binding of antigenic determinants in synthesized VLPs with antibodies to SARS-CoV-2 proteins has been demonstrated. The immunogenic properties of VLPs have been studied.
Conclusion. The production and purification of recombinant VLPs consisting of full-length SARS-CoV-2 proteins with a universal set of surface antigens have been developed and optimized. Self-assembling particles that mimic the coronavirus virion induce a specific immune response against SARS-CoV-2.
175-186
SHORT COMMUNICATION
Seroprevalence of herpes simplex virus type 1 (Herpesviridae: Simplexvirus: Human alphaherpesvirus 1) in smokers
Abstract
Introduction. Herpes simplex virus type 1 (HSV-1) is one of the most common human viral infections and has a double-stranded DNA genome belonging to the Herpesviridae family. Smoking is one of the leading causes of disease and premature death worldwide, responsible for the death of up to six million people annually. The purpose of the current study was to determine the seroprevalence of HSV-1 infection among smokers.
Methods. The search strategy was conducted in the period from December 2022 to January 2023. The study included a random sample of 94 (88 males, and 6 females) healthy participants, aged between ≤ 20 to ≥ 60 years, with 50 participants as the control group. The HSV serological testing consisted of detecting antibodies to HSV-1 IgG with the help of ELISA.
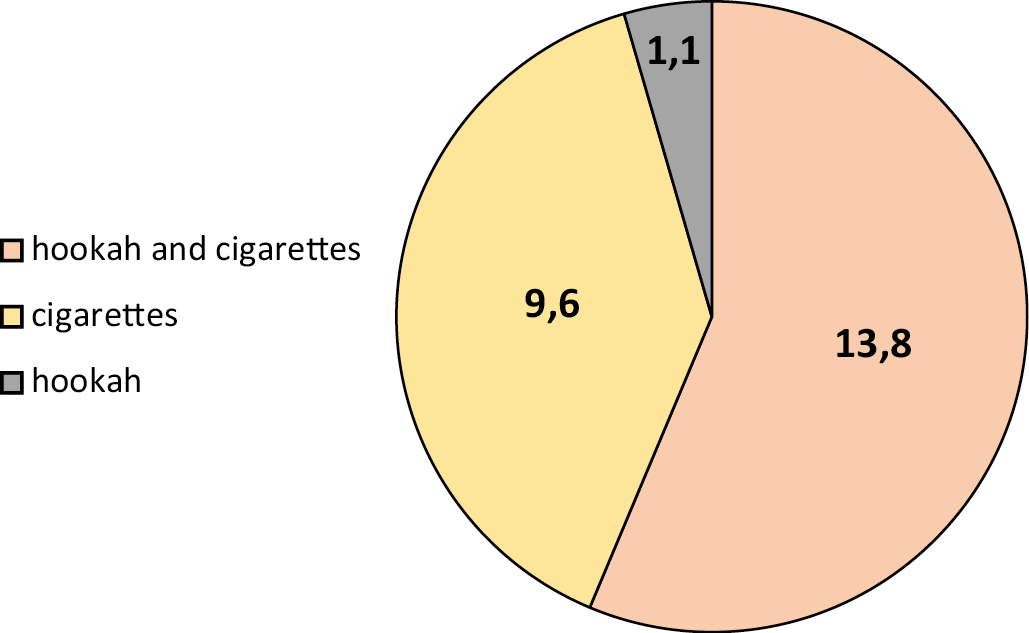
Results. Most participants were university students, consisting of 45.7% males and 5.3% females, followed by employed smokers, consisting of 0.2% males and 1.1% females. The number of females was much lower than that of males reaching 6.4 and 93.6% respectively, due to customs and traditions. The seroprevalence was 24.47, 22.3 and 2.1% in males and females respectively. The seroprevalence rate was 13.8% in hookah and cigarette smokers, 9% in cigarette smokers and 1.1% in hookah smokers exclusively. The highest rate was observed in the age groups of 21-30 and 31–40 years with 12.80% and 7.40% respectively.
Conclusions. The study revealed that the seroprevalence of HSV-1 IgG was 24.47%, and was higher among hookah and cigarette smokers compared to those who exclusively smoked cigarettes or hookah.
187-192
ANNIVERSARY DATES
An outstanding figure in domestic healthcare and medical science of the twentieth century, Viktor Mikhailovich Zhdanov
 193-194
193-194
Регистрационный номер и дата принятия решения о регистрации СМИ: серия ПИ № ФС77-77676 от 29.01.2020.