



Phylogenetic analysis of variants of the Puumala virus (Hantaviridae: Orthohantavirus) circulating in the Saratov region
- Authors: Krasnov Y.M.1, Naidenova E.V.1, Guseva N.P.1, Polunina T.A.1, Sharapova N.A.1, Sosedova E.A.1, Kotova N.V.1, Zakharov K.S.1, Kazantsev A.V.1, Domanina I.V.1, Chekashov V.N.1, Shilov M.M.1, Kondratiev E.N.1, Osina N.A.1, Kutyrev V.V.1
-
Affiliations:
- Russian Research Anti-Plague Institute «Microbe»
- Issue: Vol 69, No 2 (2024)
- Pages: 162-174
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16616
- DOI: https://doi.org/10.36233/0507-4088-224
- EDN: https://elibrary.ru/ijmubu
- ID: 16616

Cite item

Abstract
The objective is to determine the complete nucleotide sequence and conduct a phylogenetic analysis of genome variants of the Puumala virus isolated in the Saratov region.
Materials and methods. The samples for the study were field material collected in the Gagarinsky (formerly Saratovsky), Engelssky, Novoburassky and Khvalynsky districts of the Saratov region in the period from 2019 to 2022. To specifically enrich the Puumala virus genome in the samples, were used PCR and developed a specific primer panel. Next, the resulting PCR products were sequenced and the fragments were assembled into one sequence for each segment of the virus genome. To construct phylogenetic trees, the maximum parsimony algorithm was used.
Results. Genetic variants of the Puumala virus isolated in the Saratov region have a high degree of genome similarity to each other, which indicates their unity of origin. According to phylogenetic analysis, they all form a separate branch in the cluster formed by hantaviruses from other subjects of the Volga Federal District. The virus variants from the Republics of Udmurtia and Tatarstan, as well as from the Samara and Ulyanovsk regions, are closest to the samples from the Saratov region.
Conclusion. The data obtained show the presence of a pronounced territorial confinement of strains to certain regions or areas that are the natural biotopes of their carriers. This makes it possible to fairly accurately determine the territory of possible infection of patients and/or the circulation of carriers of these virus variants based on the sequence of individual segments of their genome.
Full Text
Introduction
It is known that the Volga Federal District (VFD) of the Russian Federation accounts for more than 80% of all annually registered cases of hemorrhagic fever with renal syndrome (HFRS) [1, 2], and the Saratov region is no exception. Thus, according to the data presented in the letter of the Chief State Sanitary Doctor of the Russian Federation A.Yu. Popova No. 02/4030-2024-32 dated 11.03.2024 «On the epidemiological situation of hemorrhagic fever with renal syndrome in 2023 and forecast for 2024», in 2023 in Russia 5093 cases of HPRS were registered (3.47 per 100 thousand of the population; the average annual rate (2012–2023, excluding 2020 and 2021) was 5.34 per 100 thousand of the population), 84.7% of which were in the Volga Federal District.
Zoning of the territory of the Saratov region showed that in 16 out of 38 administrative districts for the 2010–2022 period, there was a high intensity of HFRS epidemic manifestations. Among the most unfavorable are the Gagarinsky district (formerly Saratovsky district), as well as the Kumysnaya Polyana natural park, located in the city of Saratov. The area of the park is 4417 hectares [3]. As part of the organization of local government, the territory of the Gagarinsky district from January 1, 2022 is included in the urban district of the city of Saratov, while not being part of the city limits.
In the European part of Russia, including the Saratov Region, the most common causative agent of HFRS is Puumala virus (Puumala orthohantavirus), the main natural host of which is the red vole (Myodes glareolus Schreber, 1780) [4, 5]. Puumala virus, like other hantaviruses, belongs to the Orthohantavirus genus of the Hantaviridae family [6]. The viral genome is represented by segmented single-stranded RNA of negative polarity. The large segment of the genome (L) encodes the viral RNA polymerase RdRp, the medium segment (M) encodes two surface glycoproteins G1 and G2, and the small segment (S) encodes the nucleocapsid protein (N) [7].
In a previous study [8], together with colleagues from the State Scientific Center of Virology and Biotechnology «Vector», we sequenced the complete genomes (segments S, M and L) of three samples taken from red voles caught in the Kumysnaya Polyana natural park (Saratov) in mid-2019. Analysis of the results obtained confirmed that it was Puumala virus that caused the outbreak of HFRS observed in Saratov at that time. The nucleotide sequences obtained were close to the previously described variants from the Republics of Tatarstan and Udmurtia, as well as the Samara region. There was conducted a comparative phylogenetic analysis of Puumala virus variants circulating in red voles captured in the Republic of Tatarstan [9, 10], and comparison with the samples from the Republic of Udmurtia and the Ulyanovsk Region [11]. The analysis showed the similarity of virus genomes from the studied regions with the variants isolated in Tatarstan. There was demonstrated that the identified Puumala virus isolates from the Tyumen, Arkhangelsk, and Omsk regions form a common Eastern Finnish branch, which raises additional questions about the distribution routes of the main carrier. Furthermore, data are presented comparing genenic variants from Tatarstan, Udmurtia, Samara, and Saratov. The authors conclude that Puumala virus variants are likely to have arisen in these regions as a result of reassortment, since they contained segments S and L belonging to the Bashkir branch and segment M probably derived from sublineage ancestors from Kursk, Moscow and Ivanovo regions [12]. An attempt was also made to model the phylogeographic distribution of Puumala hantavirus throughout Europe during the last postglacial period [13].
Thus, the aim of the presented study is to determine the complete nucleotide sequence of the genome of various variants of Puumala virus isolated in the Saratov region and to conduct phylogenetic analysis of the obtained data.
Materials and methods
The samples for the study were organ samples of small mammals, mainly red vole, collected in 2019–2022 in the suburban and urban area of Saratov (Kumysnaya Polyana natural park), as well as in the Gagarinsky, Engelssky, Novoburassky and Khvalynsky districts of the Saratov region. The zoological group made regular visits to the points determined by the annual monitoring plan, where the capture was carried out.
Authors confirm compliance with institutional and national standards for the use of laboratory animals in accordance with «Consensus author guidelines for animal use» (IAVES 23 July 2010). The study was approved by the Bioethics Commission of the Russian Anti-Plague Institute «Microbe» (protocol No. 8, dated 21.11.2023).
Nucleic acids were isolated using the AmpliPrime RIBO-prep RNA/DNA extraction kit, and reverse transcription was performed with the Reverta-L reagent kit (Central Research Institute of Epidemiology, Russian Federation). The presence of genetic material of hantaviruses was determined by RT-PCR with the OM-Screen-HPRS-RT reagent kit (Syntol Research and Development Company, Russian Federation).
A panel of 38 samples of small mammal lung suspensions in which Puumala virus RNA was detected by RT-PCR was compiled for the study. For specific enrichment of the viral genome (S, M, and L segments) in samples from field material, a set of primers calculated by the authors was used, which is listed in Table 1.
Table 1. Oligonucleotide primer used in the work
Таблица 1. Последовательности олигонуклеотидных праймеров, используемых в работе
Primer Праймер | Primer sequence Структура праймера | Annealing T, °C Температура отжига, °C | Amplicon length, bp Длина ампликона, п.н. |
Segment S Сегмент S | |||
Pum_S_1f | TAGTAGTAGACTCCTTGAAAAAGCTAC | 50 | 370 |
Pum_S_1r | CAATGTCAATGGCGTTCAC | ||
Pum_S_2f | GAAGAATGGCAGATGCTGTGTCCC | 58 | 454 |
Pum_S_2r | ACGGTCTGTCTTCCACGAGTTGAC | ||
Pum_S_3f | CGACTGGGATTGAACCTGATGATC | 56 | 344 |
Pum_S_3r | CGGGTGTAAGTTCCTCAGCTTTC | ||
Pum_S_4f | CATTTGAGGATATTAATGGCTTTAGG | 54 | 376 |
Pum_S_4r | AATCAACTTATCAATGTCTGCCAC | ||
Pum_S_5f | TGCGTAATATCATGAGTCCAGTGATG | 56 | 355 |
Pum_S_5r | AGCCATCCCAGCAACATAAATG | ||
Pum_S_6f | ACATCGAATCTCCTAATGCACC | 54 | 354 |
Pum_S_6r | TGATCTATGAGTGACTGAGCAAGG | ||
Pum_S_7f | TGCTCAGTCACTCATAGATCAGAAAG | 53 | 400 |
Pum_S_7r | AGCTCAGTTTCACATTCTTGGG | ||
Pum_S_7-2f | AATCAGGAGCCCTTAAAGATATG | 53 | 467 |
Pum_S_7-2r | TCAGCATGTTGAGGTAGTATGTTGTG | ||
Pum_S_fin_f | GTTTTGAATTAATGCACTAATCAGGG | 50 | 350 |
Pum_S_fin_r | TAGTAGTATGCTCCTTGAAAAGCAATC | ||
Segment M Сегмент М | |||
Pum_М_1f | TAGTAGTAGACTCCGCAAGAAGAAGC | 54 | 386 |
Pum_М_1r | ATCCTCTCAAATTCACTTCACTGC | ||
Pum_М_2f | CATGGGAAATTAAAGGTGATCTTG | 54 | 309 |
Pum_М_2r | AATTGCCCTGAAACACAGTATG | ||
Pum_М_3f | GTTTGATCCCTACTTTAGTGGTTG | 53 | 349 |
Pum_М_3r | CTTCATACTATCACCAGATGTCACC | ||
Pum_М_4f | ATACTGTGTTTCAGGGCAATTGG | 56 | 386 |
Pum_М_4r | CCTGTTACTTTCCCAGCAATACGTAG | ||
Pum_М_5f | ATCGTTCTGCTGAAGTTCTTTCAAG | 56 | 345 |
Pum_М_5r | AAGACTGTGCATTGTGTTGTCTTCTC | ||
Pum_М_6f | ACTGGGTTCATCTCATTACCTGG | 54 | 341 |
Pum_М_6r | TAACTCTACGGCAAGAGAGTGTG | ||
Pum_М_7f | GAGTTATGTGTACCAGGTCTTCACGG | 55 | 340 |
Pum_М_7r | GCTTGAAGGGCAGATGTTGTTG | ||
Pum_М_8f | ATGGGCTCGATGGTCTGTGAGG | 58 | 380 |
Pum_М_8r | TATCCCAGACCCGTGTGCTGTGTC | ||
Pum_М_9f | CTCTCTTTAGGTATCGGAGTCGG | 55 | 392 |
Pum_М_9r | CAGCAGTTTGCCAAGGATAAGC | ||
Pum_М_10f | AAACTGCATTTCATTGCTATGGTTC | 55 | 326 |
Pum_М_10r | TTGAAATTGTCCCTATCAAACACAC | ||
Pum_М_10-2f | GGCTGTAACCCACTTGATTGCC | 58 | 430 |
Pum_М_10-2r | CCATCAAACTGGCACACAGGTGTTG | ||
Pum_М_11f | TAATATTTAAGCAATGGTGCACTAC | 53 | 452 |
Pum_М_11r | CCTTAATTGAAGTAAGAAATGCAG | ||
Pum_М_12f | GATGGGAATACAATTTCAGGATAC | 53 | 307 |
Pum_М_12r | GCCTTAATTGAAGTAAGAAATGCAG | ||
Pum_М_13f | GAATGGATTGATCCTGACAGTTCAC | 56 | 502 |
Pum_М_13r | ATCCAATTTCCATTGAGGACCC | ||
Pum_М_13-2f | CAATGTGTTACGGATCTACTACAGC | 54 | 393 |
Pum_М_13-2r | GCAACTATCTACTAAGGCTTATGCTC | ||
Pum_М_fin_f | GACAGGATACAACCAAGCAGATAGTG | 54 | 417 |
Pum_М_fin_r | TAGTAGTAGACTCCGCAAGAACAAAAG | ||
Segment L Сегмент L | |||
Pum_L_1f | TAGTAGTAGACTCCGAGATAGAGAAG | 51 | 468 |
Pum_L_1r | TGTGCTTCTACCTGTAGTTGTTGCTC | ||
Pum_L_2f | ATGATGTGATACAAAGCATGGAG | 53 | 453 |
Pum_L_2r | TCCTAAAGCCAGATTGACAATTAC | ||
Pum_L_3f | GTTTATTGAGCAACAACTACAGGTAG | 53 | 394 |
Pum_L_3r | GCCCAGTTACTTCTTTAAATGC | ||
Pum_L_4f | TGTAAGAATTGGCTCGGAACTGATC | 56 | 421 |
Pum_L_4r | TCAATCAATGCCTTCGACTTAGGATC | ||
Pum_L_5f | ATCTCAAAGGATTCAAAGAAAGGG | 54 | 391 |
Pum_L_5r | TCAAGTGATTTAGATGGCAGGATAC | ||
Pum_L_6f | CGTGATATTACTGAATCTCTTATTGC | 53 | 406 |
Pum_L_6r | TGAGATGTGGAAAGAAAGAAATGG | ||
Pum_L_7f | CTTCTTGCTACAGCTACATGGTTTC | 54 | 410 |
Pum_L_7r | ACGATAATGCTTATACACGACCC | ||
Pum_L_8f | AAGACACTATTAGTTAGCTTAGCCC | 52 | 354 |
Pum_L_8r | GCTGTTCAACAACTACCTGATTG | ||
Pum_L_9f | TTCATCTTGAAACAGTTGAATGGG | 54 | 438 |
Pum_L_9r | CTTTGGTATTTCCTAACAATTCTTGC | ||
Pum_L_9-2f | TCATCTTGAAACAGTTGAATGGG | 54 | 341 |
Pum_L_9-2r | AAACGAATTGCCTCAATGAGAG | ||
Pum_L_10f | GTCAGGAGCATTACAAGAAGATGGTC | 56 | 369 |
Pum_L_10r | CAACGAGGACTGGATTTCACTTTCTC | ||
Pum_L_11f | ATTCAACAGGCTTTAGAGAAGGC | 54 | 450 |
Pum_L_11r | GATATAATTTGGCCCACACACG | ||
Pum_L_12f | AATATGGGTGAATTGTCTGATGAAG | 54 | 440 |
Pum_L_12r | GAATGTAGATAGAAACTCCGCATTTG | ||
Pum_L_13f | GGCAGGAAACTTTCATTGGCATG | 57 | 440 |
Pum_L_13r | GACATAGAACCATCACCACCTAACGG | ||
Pum_L_14f | TTATATGGTACTGCTCCTGGTATGG | 54 | 383 |
Pum_L_14r | AGGATGTTGTTCACTCCAAAGCTC | ||
Pum_L_15f | TTTGTAGGTAAAGTTCAGTGGAAAG | 53 | 459 |
Pum_L_15r | TTGGTCTATGAATTTGTCTTGTTG | ||
Pum_L_15-2f | CTTTGGAGTGAACAACATCCTGTC | 54 | 320 |
Pum_L_15-2r | ATCCCTCCAGGCATATTCTTTAG | ||
Pum_L_16f | GGATATGGAGCTCTTTCAAACACTTG | 55 | 387 |
Pum_L_16r | CACAATTACAATCCTCGACTTTCC | ||
Pum_L_17f | TTATTGGTTGAAGACTATGTCTCTTG | 52 | 441 |
Pum_L_17r | ATTAAATGTTACCCTCAAATCTCC | ||
Pum_L_18f | CTTTATGAAGGAGATTTGAGGGTAAC | 53 | 423 |
Pum_L_18r | CCTTTAGATTATGGTATGCATGG | ||
Pum_L_19f | CTAATGGATTTAGGGCTATGGC | 54 | 385 |
Pum_L_19r | TGACCATTGAGTACTAGAGATTGTGC | ||
Pum_L_20f | AGCACAATCTCTAGTACTCAATGGTC | 53 | 405 |
Pum_L_20r | CGAACTCTGTTAAATCATACGGATC | ||
Pum_L_fin_f | GTCCAAGCATTACAATTTCCATAC | 52 | 304 |
Pum_L_fin_r | TAGTAGTATGCTCCGAGAAAAGAGC | ||
Further sequencing of the obtained PCR products was performed using a genetic analyzer (AB 3500xl and/or Ion S5), according to the recommendations of the manufacturer of this equipment (Thermo Fisher Scientific, USA). MEGA7 (http://www.megasoftware.net/) and UGENE (http://ugene.net) software were used to assemble sequenced fragments into a single sequence.
When constructing phylogenetic trees, the sequence of segment S (the central part of 1071 nucleotides), as well as most of the sequence of segments M and L of the Puumala virus genome, represented in the international database NCBI GeneBank, were used. This section of the work was performed using the maximum parsimony algorithm in the BioNumerics 7.6 software package (Applied Maths, Belgium).
Results and discussion
As a result of this study, nucleotide sequences of the genome of Puumala virus circulating in the Saratov region were obtained, 33 of which with the highest quality reads were deposited in the international database NCBI GenBank (Table 2).
Table 2. List of Puumala orthohantavirus sequences obtained in this study and deposited in the NCBI GenBank database
Таблица 2. Нуклеотидные последовательности вируса Пуумала, полученные в результате исследования
No. № | Sample Образец | Place of the sample collection Место получения образца | Year of collection Год сбора материала | GenBank Accession Number Номер в базе NCBI GenBank | ||
segment S сегмент S | segment M сегмент M | segment L сегмент L | ||||
1. | 114 | Gagarinsky district Гагаринский район | 2019 | OL343591.1 | OL343569.1 | OL343547.1 |
2. | 131 | 2019 | OL343593.1 | OL343571.1 | OL343549.1 | |
3. | 296 | 2019 | OL343594.1 | OL343572.1 | OL343550.1 | |
4. | 348 | 2019 | OL343595.1 | OL343573.1 | OL343551.1 | |
5. | 420 | 2019 | OL343596.1 | OL343574.1 | OL343552.1 | |
6. | 422 | 2019 | OL343597.1 | OL343575.1 | OL343553.1 | |
7. | 439 | 2019 | OL343598.1 | OL343576.1 | OL343554.1 | |
8. | 525 | 2019 | OL343599.1 | OL343577.1 | OL343555.1 | |
9. | 645 | 2019 | OL343600.1 | OL343578.1 | OL343556.1 | |
10. | 656 | 2019 | OL343601.1 | OL343579.1 | OL343557.1 | |
11. | 696 | 2019 | OL343602.1 | OL343580.1 | OL343558.1 | |
12. | 701 | 2019 | OL343603.1 | OL343581.1 | OL343559.1 | |
13. | 836 | 2019 | OL343604.1 | OL343582.1 | OL343560.1 | |
14. | 988 | 2019 | OL343605.1 | OL343583.1 | OL343561.1 | |
15. | 989 | 2019 | OL343606.1 | OL343584.1 | OL343562.1 | |
16. | 1039 | 2019 | OL343585.1 | OL343563.1 | OL343541.1 | |
17. | 1042 | 2019 | OL343586.1 | OL343564.1 | OL343542.1 | |
18. | 1059 | 2019 | OL343587.1 | OL343565.1 | OL343543.1 | |
19. | 1081 | 2019 | OL343588.1 | OL343566.1 | OL343544.1 | |
20. | 1107 | 2019 | OL343589.1 | OL343567.1 | OL343545.1 | |
21. | 1109 | 2019 | OL343590.1 | OL343568.1 | OL343546.1 | |
22. | 1186 | 2019 | OL343592.1 | OL343570.1 | OL343548.1 | |
23. | 3 | 2022 | OQ032670.1 | * | * | |
24. | 29 | 2022 | OQ032672.1 | * | * | |
25. | 30 | 2022 | OQ032673.1 | * | * | |
26. | 31 | 2022 | OQ032671.1 | * | * | |
27. | 99 | Engelssky district Энгельсский район | 2022 | OQ032667.1 | * | * |
28. | 100 | 2022 | OQ032669.1 | * | * | |
29. | 101 | 2022 | OQ032668.1 | * | * | |
30. | 117 | Khvalynsky district Хвалынский район | 2021 | OR999067 | * | * |
31. | 122 | 2021 | OR999068 | * | * | |
32. | 129 | 2021 | OR999069 | * | * | |
33. | 272 | Novoburassky district Новобурасский район | 2021 | OR999070 | * | * |
Note. * – Data at the analysis stage.
Примечание. * – данные на этапе анализа.
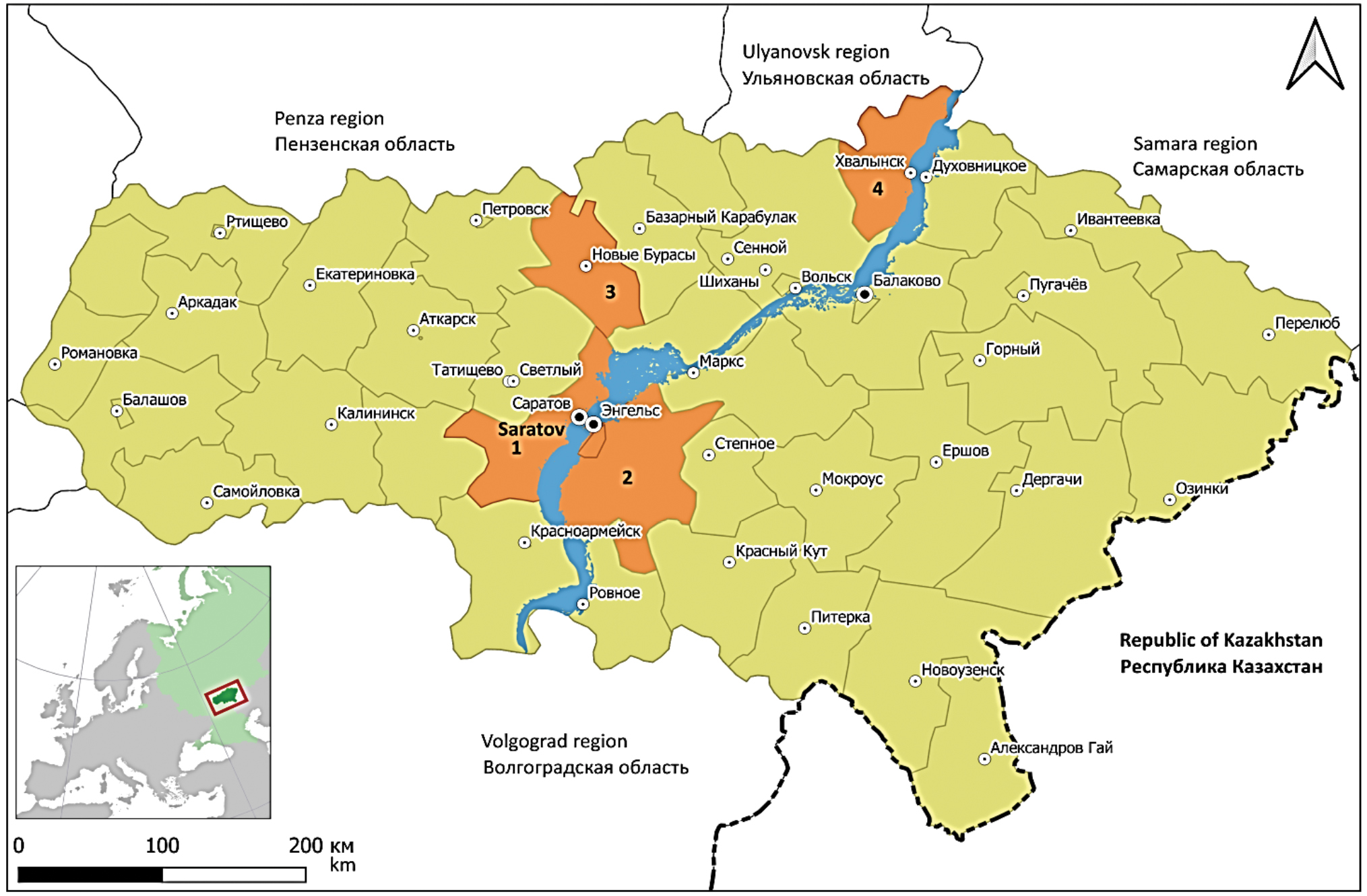
A comparative analysis of 26 nucleotide sequences of the Puumala virus whole genome from suburban and urban areas of Saratov (Gagarinsky district) showed their high similarity with each other (98.6-100%), regardless of the year of isolation. Samples from Novoburassky (No. 272) and Engelssky (Nos. 99, 100, 101) districts have genome similarity of 98.4–99.1% with 26 samples from Gagarinsky district. Three samples from the Khvalynsky district, Saratov region (Nos. 117, 122, 129) have 95.5% similarity with all variants from the Gagarinsky district. Thus, a rather large number of differences among the variants of the virus genome from the Khvalynsky district was revealed compared to samples from the Engelssky, Gagarinsky and Novoburasky districts bordering each other (Fig. 1).

Fig. 1. Administrative division of the Saratov region. The city district of Saratov (1), Engelssky (2), Novoburassky (3) and Khvalynsky (4) districts are highlighted in color.
Рис. 1. Административное деление Саратовской области. Цветом выделены Гагаринский район и городской округ Саратова (1), Энгельсский (2), Новобурасский (3) и Хвалынский (4) районы.
Using the available nucleotide sequences of the S segment of the Puumala virus genome presented in the international genetic database, an analysis including 368 viral variants was performed (Fig. 2 a) and a general phylogenetic tree was constructed. Comparison of sequences of M or L segments of the hantavirus genome is limited by their much smaller number relative to the S segment represented in the database, but the clustering pattern changes insignificantly, retaining, as a rule, the same mutual arrangement.
When phylogenetic relatedness was established, the S segment of the Puumala virus genome variants from the Saratov region formed a common cluster with viral sequences from the Ulyanovsk, Samara, and Penza regions, as well as from the Republics of Tatarstan, Bashkortostan, and Udmurtia. Sequence variants of the same genome segment obtained in other Russian territories (Omsk, Tyumen, Arkhangelsk, Moscow, Ivanovo and Kursk regions, and the Republic of Karelia) were significantly distant from the cluster formed by sequences from the above-mentioned subjects of the Volga Federal District. It is worth noting that there are two samples (NCBI, MT822195 and MT822196) on the same branch formed by genomes from Samara region, obtained from clinical material collected in January 2020 in Switzerland (Fig. 2 a, b). But the annotation of these sequences does not provide information about the possible site of infection of HPRS patients, making it difficult to draw clear conclusions. Otherwise, the cluster formed by viral sequences (segment S) from the territory of the Volga Federal District did not include any variants from neither the other Federal Districts of Russia, nor any nearby or far abroad countries.
Within cluster 1 (Fig. 2 a, b), formed by Puumala virus genomes from samples from the territory of the Volga Federal District, the degree of sequence similarity is 91.4–100%. Despite the fact that only the central part of the S segment sequence (1071 bp) was used in the comparative analysis, each variant from this cluster has at least 62 unique single mutations that are common to all samples from cluster 1 and distinguish these variants from all others (Fig. 2 a, b). Figure 2 b shows that variants of the Puumala hantavirus genome from the Saratov urban district and Engelssky district, and one from Novoburassky district, form a compact group in the form of a separate branch. The places where carriers of the Puumala virus were captured from Udmurtia within the urban district of Saratov were separated from each other from several hundred meters to 9 km. Between the places of capture of Puumala virus carriers within the urban district of Saratov and Engelssky district, the distance in a straight line is 14–19 km, and for a sample from the Novoburassky district – about 46 km. Despite the significant remoteness, a high degree of similarity (98% or more) is observed for Puumala virus genome variants from these territories, which may indicate a single biotope of the main carriers and their free movement within these regions. The Khvalynsky district is located in the north of the Saratov region (more than 100 km distance of from the other regions studied) and borders on the Ulyanovsk and Samara regions (Fig. 1). This fact is clearly reflected in the location of Puumala virus variants in cluster 1 (Fig 2 b). There was demonstrated that the samples obtained from Khvalynsky district and variants from Gagarinsky, Engelssky and Novoburassky districts are on the different branches differing from them by 55–78 single nucleotide polymorphisms (SNPs) and have similarity with the latter of 92.7–96.9%. The closest to hantavirus variants from the Khvalynsky district are the genomes of Puumala virus from the Samara region, followed by its variants from the Republic of Tatarstan and Ulyanovsk region (Fig. 2 b).

Fig. 2. Phylogenetic tree showing the close relationship between 368 genome variants (segment S) strains of the Puumala orthohantavirus from Russia, near and far abroad countries. a – for three clusters; b – enlarged fragment of the phylogenetic tree – cluster 1 in Figure 2 a. The close relationship between genome variants (segment S) of 151 strains of the Puumala orthohantavirus from the Volga Federal District is shown. In Figure 2 b, samples circled in red remained the only variants from the republics of Bashkortostan and Tatarstan, Samara and Ulyanovsk regions in the case of phylogenetic trees for segments M and L.
Рис. 2. Филогенетическое дерево, показывающее близость родства между 368 вариантами генома (сегмент S) вируса Пуумала с территории России, стран ближнего и дальнего зарубежья. а – по трем кластерам; б – увеличенный фрагмент филогенетического дерева – кластер 1 на рис. 2 а. На рис. 2 б образцы, обведенные красным цветом, остались единственными вариантами из республик Башкортостан и Татарстан, Самарской и Ульяновской областей в случае филогенетических деревьев по сегментам M и L.
A detailed examination of the location of genome variants (S segment) of Puumala virus from Saratov region (Fig. 2 b) shows very close affinity between samples from Gagarinsky district, as well as their great similarity with samples from the Engelssky and Novoburassky districts. The difference between the sequence of the S segment (1071 bp) of the Puumala virus variant isolated in August 2021 in the Novoburassky district and the closest Puumala variant obtained in May 2019 in the territory of the Gagarinsky district is 5 SNPs. The differences between the sequences of the S segment (1071 bp) of the Puumala virus variant detected in July 2022 in the Engelssky district from the closest one obtained in February 2020 during the survey of the Gagarinsky district are 15 SNPs. Between all investigated variants of Puumala virus circulating in the Gagarinsky district of Saratov region, the maximum difference in the sequence of the S segment (1071 bp) is 22 SNPs (98% similarity or more).
Molecular clock analysis performed by researchers [14, 15] showed that the S segment of the hantavirus genome evolves at a rate of about 6.7–10−4 single substitutions per year. These data suggest a rather slow change rate in the genome of Puumala virus in nature. In general, clustering by the S segment shows a common origin from one ancestral virus, or several close variants of it, for all samples from the territory of the Volga Federal District used in the analysis.
Comparative phylogenetic analysis of Puumala virus by M or L segments of samples obtained in the territory of the Saratov region shows certain changes in clustering, however, preserving the general trends of mutual arrangement.
It is worth noting that for segments M or L there is significantly less data on the genome sequences of the Puumala virus than for segment S. In Figure 2B, samples circled in red remained the only variants from the republics of Bashkortostan and Tatarstan, Samara and Ulyanovsk regions in the case of phylogenetic trees for segments M and L.

Fig. 3. Phylogenetic tree showing the close relationship between 94 genome variants (segment M) of Puumala orthohantavirus strains from Russia, near and far abroad countries.
Note: Bashkortostan (NCBI: AB297666, AF442614, KT885051, L08754, MH251332, MK496160, MZ673553, NC_077666); Kursk region (MZ580943, MZ580946, MZ580949, MZ580952); Ivanovo region (OP561838); Moscow region (OP561841); Samara region (AB433850, AB433852); Ulyanovsk region (OP561826); Tatarstan (Z84205); Udmurtia (OP561835); Penza region (OP561847); Switzerland (MT822194); Omsk region (AF367061, AF442615, AF442616, AF442617); Arkhangelsk region (OP561850); Tyumen region (OP561853).
Рис. 3. Филогенетическое дерево, показывающее близость родства между 94 вариантами генома (сегмент М) вируса Пуумала с территории России, стран ближнего и дальнего зарубежья.
Примечание: Башкортостан (AB297666, AF442614, KT885051, L08754, MH251332, MK496160, MZ673553, NC_077666); Курская обл. (MZ580943, MZ580946, MZ580949, MZ580952); Ивановская обл. (OP561838); Московская обл. (OP561841); Самарская обл. (AB433850, AB433852); Ульяновская обл. (OP561826); Татарстан (Z84205); Удмуртия (OP561835); Пензенская обл. (OP561847); Швейцария (MT822194); Омская обл. (AF367061, AF442615, AF442616, AF442617); Архангельская обл. (OP561850); Тюменская обл. (OP561853).
Figure 3 shows the result of phylogenetic clustering between 94 genome variants (M segment, 2920 bp locus) of Puumala virus from the territory of Russia, near and far abroad. The phylogenetic tree obtained shows that the samples from the Gagarinsky district (Saratov ar.) still form a homogeneous group and are in the same cluster with the variants from the Volga Federal District, while the closest in similarity to them are the virus variants from the Republics of Udmurtia (OP561835, similarity of 94.9–95.6%), Tatarstan (Z84205, similarity of 94.6–95.3%) and Samara region (AB433850, similarity of 91.3–92.0% and AB433852, similarity of 90.6–91.3%). It should be noted that when clustering by M segment, the virus genome sequences obtained from Moscow, Kursk, and Ivanovo regions are closer to the variants from the territory of the Volga Federal District than the variants from the Republic of Bashkortostan, which is part of the Volga Federal District. In the M segment, variants from Bashkiria form a separate branch that is not included in clusters 1 and 2. In contrast, during phylogenetic clustering of the virus by S segment, all sequences from the Volga Federal District were in the same cluster and significantly distant from variants from Central Russia. It is likely that the hantavirus obtained in the Republic of Bashkortostan had reassortment along the M segment with some variant of this virus from a region that is not part of the Volga Federal District.
Some authors’ conclusion [12] that variants of Puumala virus likely arose in Tatarstan, Udmurtia, Samara, and Saratov as a result of reassortment, since they contained segments S and L belonging to the Bashkir branch and segment M derived from sublineage ancestors from Kursk, Moscow, and Ivanovo regions, was not confirmed in this study. Stable preservation of the unity of phylogenetic isolation of Puumala virus genomes on the territory of the subjects of the Volga Federal District is evident, with the exception of variants from the territory of Bashkortostan for segment M (Fig. 3) and a sample from the Penza Region for segment L (Fig. 4). In Figure 3, which shows the M segment affinity, any of the variants of the Puumala virus genome in cluster 1 differs by at least 433 SNPs (similarity of 85.2%) from any of the variants of the Puumala virus genome in cluster 2. Variants from Bashkortostan differ by at least 531 SNPs (similarity of 81.8%) from any of the variants in both cluster 1 and cluster 2. The branch with the closest variants of Puumala virus from Finland has about 580 SNPs (similarity of 80.1%) from variants from Bashkortostan.
The lack of data from most regions of Russia on Puumala virus variants circulating in them makes it very difficult to draw conclusions about possible pathways of reassortant emergence.

Fig. 4. Phylogenetic tree showing the close relationship between 76 genome variants (segment L) of Puumala orthohantavirus strains from the territory of Russia, near and far abroad countries.
Note: Bashkortostan (NCBI: AB297667, KT885050, MK496161, MK496164, MH251333, NC_077667); Samara region (AB574183, AB574184); Ulyanovsk region (OP561825); Udmurtia (OP561834); Tatarstan (EF405801); Switzerland (MT822193); Kursk region (MZ580944, MZ580947, MZ580953, MZ580950); Moscow region (OP561840, OP561843); Ivanovo region (OP561837); Penza region (OP561846); Arkhangelsk region (OP561849); Tyumen region (OP561852).
Рис. 4. Филогенетическое дерево, показывающее близость родства между 76 вариантами генома (сегмент L) вируса Пуумала с территории России, стран ближнего и дальнего зарубежья.
Примечание: Башкортостан (AB297667, KT885050, MK496161, MK496164, MH251333, NC_077667); Самарская обл. (AB574183, AB574184); Ульяновская обл. (OP561825); Удмуртия (OP561834); Татарстан (EF405801); Швейцария (MT822193); курская обл. (MZ580944, MZ580947, MZ580953, MZ580950); Московская обл. (OP561840, OP561843); Ивановская обл. (OP561837); Пензенская обл. (OP561846); Архангельская обл. (OP561849); Тюменская обл. (OP561852).
Figure 4 shows the result of phylogenetic clustering between 76 genome variants (segment L, locus at 6405 bp) of Puumala virus from Russia, near and far abroad. All variants from the Gagarinsky district (Saratov ar.) of the Saratov region are invariably in the same cluster with other Puumala viruses from the Volga Federal District. The closest to the variants from Saratov region, as in the case of segment M, are Puumala virus samples from the Republics of Udmurtia (OP561834, similarity of 95.2–95.8%) and Tatarstan (EF405801, similarity of 94.5–95.1%), as well as Samara region (AB574183 и AB574184, similarity of 91.7–92.7%). When analyzing segment L, variants of Puumala virus from the Republic of Bashkortostan, although located in the same cluster (cluster 1) together with variants of this virus from other subjects of the Volga Federal District, are significantly separated from them, maximum similarity of 86.1% (about 890 SNPs compared to the closest variant from the Ulyanovsk region in this cluster). The result of phylogenetic clustering of Puumala virus genome variants by segment L shows that the sample from Penza region is not located in cluster 1 together with virus variants from the Volga Federal District, but is in cluster 2 with variants from Ivanovo, Moscow, and Kursk regions. As can be seen from Figures 2B and 3, the sample from Penza region in terms of S and M segments belongs to cluster 1, which consists only of virus variants from the Volga Federal District. Thus, a possible reassortment of the Puumala virus variant from Penza region with variants of this virus from Ivanovo, Moscow, or Kursk regions, apparently as a result of migration of the red vole, is shown by the L segment. In previously published materials, the authors noted that this genome variant could have arisen as a result of two-step recombination [12].
Conclusion
The results of phylogenetic analysis of different variants of Puumala virus from Saratov region with the sequences presented in the NCBI GenBank database allow us to draw the following conclusions:
- – all variants of Puumala virus strains circulating in the Saratov region have a high degree of similarity between genomes, which may indicate the unity of their origin;
- – genetic sequences of Puumala virus from the Khvalynsky district have significant differences from variants obtained from the central districts of Saratov region.
- – all genetic sequences of Puumala virus obtained from the urban district of Saratov, including those of genome segments S, M and L have no reassortment.
Summarizing the obtained data, we can note the presence of pronounced territorial confinement of Puumala virus strains (or rather their carriers) to certain regions or areas. This makes it possible to accurately determine the area of possible infection of patients and the habitat of carriers of these genetic variants of Puumala orthohantavirus by the sequence of viral genome segments.
The authors confirm that there is no conflict of financial/non-financial interests related to the writing of the article.
The material has been prepared for publication on the basis of the research performed within the framework of R&D 92-2-21 Development of a system for verification of the results of indication and identification of pathogens of dangerous infectious diseases of bacterial and viral nature using molecular genetic methods (2021–2023).
About the authors
Yaroslav M. Krasnov
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-4909-2394
PhD in Chemistry, leading researcher
Russian Federation, 410005, SaratovEkaterina V. Naidenova
Russian Research Anti-Plague Institute «Microbe»
Author for correspondence.
Email: katim2003@mail.ru
ORCID iD: 0000-0001-6474-3696
PhD in Biology, leading researcher
Russian Federation, 410005, SaratovNatalia P. Guseva
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0003-3763-9708
PhD in Biology, senior researcher
Russian Federation, 410005, SaratovTatyana A. Polunina
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@mail.ru
ORCID iD: 0000-0002-2234-2760
PhD in Medical sciences, senior researcher
Russian Federation, 410005, SaratovNatalya A. Sharapova
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-5289-7783
PhD in Biology, researcher
Russian Federation, 410005, SaratovEkaterina A. Sosedova
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0009-0004-4443-2646
Researcher
Russian Federation, 410005, SaratovNina V. Kotova
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-9270-523X
Researcher
Russian Federation, 410005, SaratovKirill S. Zakharov
Russian Research Anti-Plague Institute «Microbe»
Email: zaharov_ks@mail.ru
ORCID iD: 0000-0002-4726-309X
PhD in Biology, senior researcher
Russian Federation, 410005, SaratovAndrey V. Kazantsev
Russian Research Anti-Plague Institute «Microbe»
Email: andreikazancev@mail.ru
ORCID iD: 0000-0003-1790-0411
researcher
Russian Federation, 410005, SaratovIrina V. Domanina
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-4731-8089
researcher
Russian Federation, 410005, SaratovVladimir N. Chekashov
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-9593-4353
PhD in Biology, senior researcher
Russian Federation, 410005, SaratovMikhail M. Shilov
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-0083-8212
PhD in Biology, researcher
Russian Federation, 410005, SaratovEvgeniy N. Kondratiev
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-7508-4355
Researcher
Russian Federation, 410005, SaratovNatalya A. Osina
Russian Research Anti-Plague Institute «Microbe»
Email: davidova_n_work@mail.ru
ORCID iD: 0000-0003-0954-5683
PhD in Biology, Head of Department of Microbiology
Russian Federation, 410005, SaratovVladimir V. Kutyrev
Russian Research Anti-Plague Institute «Microbe»
Email: rusrapi@mail.ru
ORCID iD: 0000-0003-3788-3452
Academician of RAS, Professor, Director
Russian Federation, 410005, SaratovReferences
- Savitskaya T.A., Ivanova A.V., Isaeva G.Sh., Reshetnikova I.D., Trifonov V.A., Ziatdinov V.B., et al. Analysis of the epidemiological situation of hemorrhagic fever with renal syndrome in the Russian Federation in 2022 and forecast of its development for 2023. Problemy osobo opasnykh infektsii. 2023; (1): 85–95. https://doi.org/10.21055/0370-1069-2023-1-85-95 https://elibrary.ru/mgxnza (in Russian)
- Ivanova A.V., Safronov V.A., Popov N.V., Kuklev E.V. Epidemiological zoning of the Volga federal district territory by the level of potential epidemic hazard of hemorrhagic fever with renal syndrome natural foci. Problemy osobo opasnykh infektsii. 2020; (1): 91–6. https://doi.org/10.21055/0370-1069-2020-1-91-96 https://elibrary.ru/jrywjk (in Russian)
- Chumachkova E.A., Ivanova A.V., Porshakov A.M., Vyatkin I.N., Forostyanaya M.V., Chumachkov K.Ya., et al. Zoning of the territory of the Saratov region by the intensity of epidemic manifestations of HFRS using GIS analysis. Problemy osobo opasnykh infektsii. 2023; (3): 156–63. https://doi.org/10.21055/0370-1069-2023-3-156-163 https://elibrary.ru/dwrlpq (in Russian)
- Tkachenko E.A., Ishmukhametov A.A., Dzagurova T.K., Bernshtein A.D., Morozov V.G., Siniugina A.A., et al. Hemorrhagic fever with renal syndrome, Russia. Emerg. Infect. Dis. 2019; 25(12): 2325–8. https://doi.org/10.3201/eid2512.181649
- Ishmukhametov A.A., Dzagurova T.K., Morozov V.G., Kurashova S.S., Balovneva M.V., Sotskova S.E., et al. Characteristics of hantaviruses as causative agents of the zoonotic hemorrhagic fevers. Epidemiologiya i vaktsinoprofilaktika. 2017; 16(3): 26–32. https://doi.org/10.31631/2073-3046-2017-16-3-26-32 https://elibrary.ru/yrhmch (in Russian)
- ICTV. Taxonomy Browser. Available at: https://ictv.global/taxonomy
- Kabwe E., Davidyuk Y., Shamsutdinov A., Garanina E., Martynova E., Kitaeva K., et al. Orthohantaviruses, emerging zoonotic pathogens. Pathogens. 2020; 9(9): 775. https://doi.org/10.3390/pathogens9090775
- Yashina L.N., Tregubchak T.V., Malyshev B.S., Smetannikova N.A., Grishchenko I.V., Dol’skii A.A., et al. Hantavirus associated with hemorrhagic fever with renal syndrome outbreak in the Saratov region in 2019. Problemy osobo opasnykh infektsii. 2021; (4): 150–6. https://doi.org/10.21055/0370-1069-2021-4-150-156 https://elibrary.ru/dxxsey (in Russian)
- Davidyuk Y.N., Kabwe E., Shamsutdinov A.F., Knyazeva A.V., Martynova E.V., Ismagilova R.K., et al. The Distribution of Puumala orthohantavirus genome variants correlates with the regional landscapes in the Trans-Kama area of the Republic of Tatarstan. Pathogens. 2021; 10(9): 1169. https://doi.org/10.3390/pathogens10091169
- Kabwe E., Shamsutdinov A.F., Suleimanova S., Martynova E.V., Ismagilova R.K., Shakirova V.G., et al. Puumala orthohantavirus reassortant genome variants likely emerging in the watershed forests. Int. J. Mol. Sci. 2023; 24(2): 1018. https://doi.org/10.3390/ijms24021018
- Kabwe E., Al Sheikh W., Shamsutdinov A.F., Ismagilova R.K., Martynova E.V., Ohlopkova O.V., et al. Analysis of Puumala orthohantavirus genome variants identified in the territories of Volga Federal District. Trop. Med. Infect. Dis. 2022; 7(3): 46. https://doi.org/10.3390/tropicalmed7030046
- Blinova E., Deviatkin A., Makenov M., Popova Y., Dzagurova T. Evolutionary formation and distribution of Puumala virus genome variants, Russia. Emerg. Infect. Dis. 2023; 29(7): 1420–4. https://doi.org/10.3201/eid2907.221731
- Castel G., Chevenet F., Razzauti M., Murri S., Marianneau P., Cosson J.F., et al. Phylogeography of Puumala orthohantavirus in Europe. Viruses. 2019; 11(8): 679. https://doi.org/10.3390/v11080679
- Souza W.M., Bello G., Amarilla A.A., Alfonso H.L., Aquino V.H., Figueiredo L.T. Phylogeography and evolutionary history of rodent-borne hantaviruses. Infect. Genet. Evol. 2014; 21: 198–204. https://doi.org/10.1016/j.meegid.2013.11.015
- Ramsden C., Holmes E.C., Charleston M.A. Hantavirus evolution in relation to its rodent and insectivore hosts: no evidence for codivergence. Mol. Biol. Evol. 2009; 26(1): 143–53. https://doi.org/10.1093/molbev/msn234
Supplementary files

