



Vol 69, No 5 (2024)

- Year: 2024
- Published: 09.11.2024
- Articles: 8
- URL: https://virusjour.crie.ru/jour/issue/view/136
Full Issue

REVIEWS
Defective HIV proviruses: possible involvement in the HIV infection pathogenesis
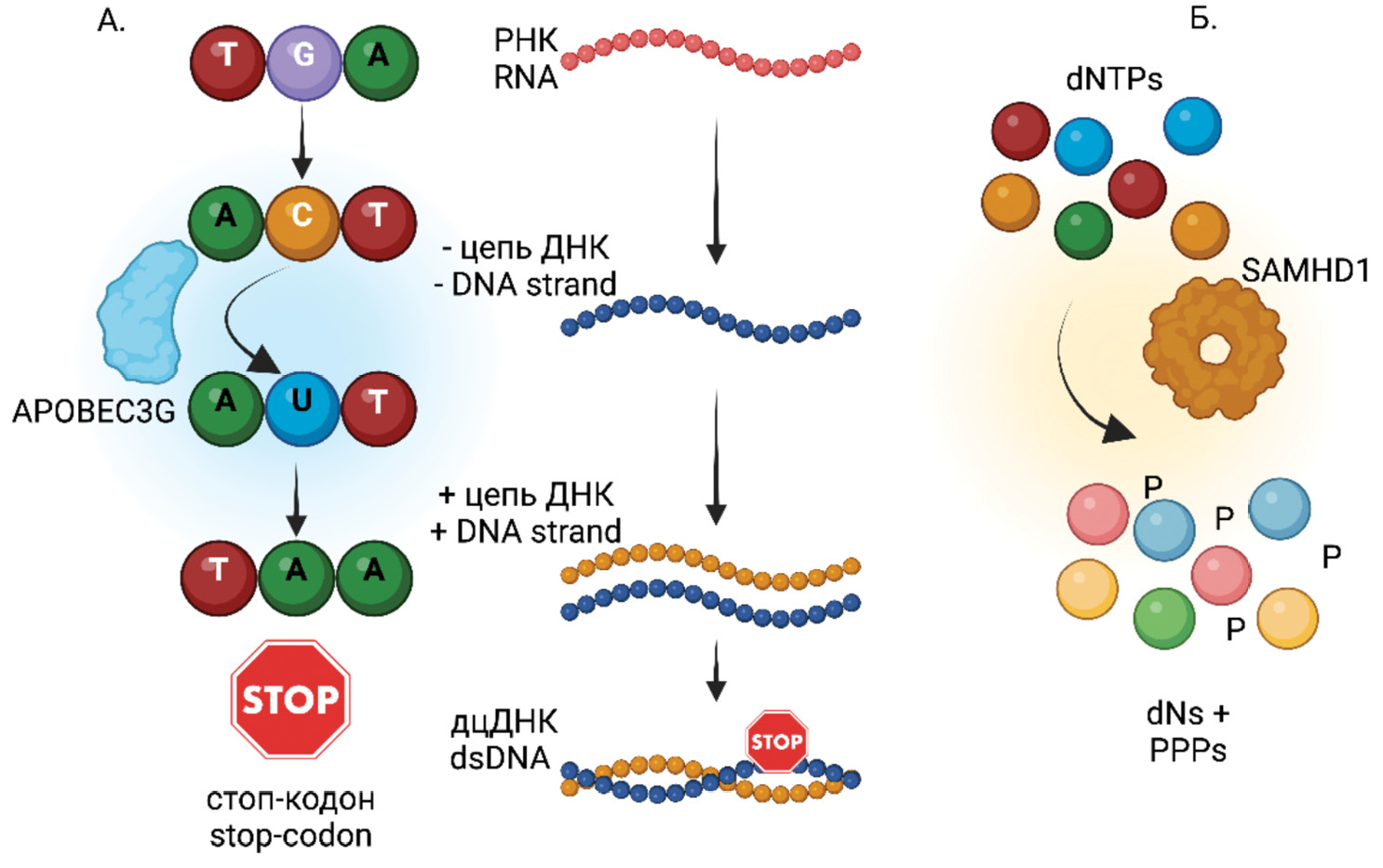
Abstract
This review article analyzes information obtained from a literature search on defective HIV genomes (HIV-1, Human Immunodeficiency Virus, Lentivirus, Orthoretrovirinae, Retroviridae). It discusses the origins of defective HIV genomes, their potential for transcription and translation, and the role of defective RNA and proteins in stimulating both innate and adaptive immunity. The article also explores their contribution to HIV pathogenesis, immune system hyperactivation despite successful antiretroviral therapy (ART), and the evolutionary processes in HIV proviral populations under ART. Additionally, it addresses challenges in reservoir elimination and HIV eradication that arise from the existence of defective HIV viruses.
 399-414
399-414



Plain-nosed bats (family Vespertilionidae) as a possible reservoir of lyssaviruses and coronaviruses in Western Siberia and the south of European Russia
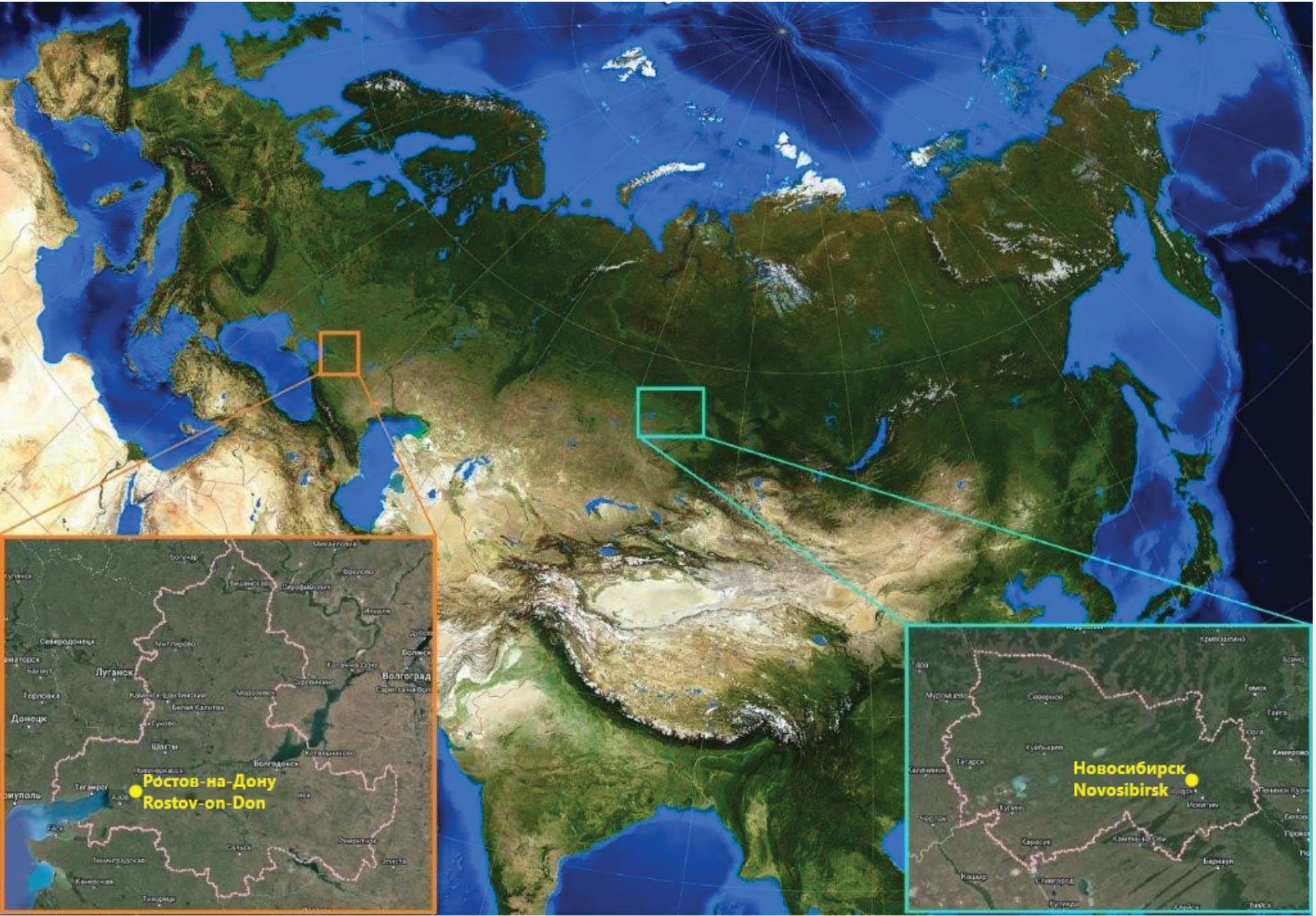
Abstract
The review presents current data on the chiropterofauna inhabiting Western Siberia and the south of the European part of Russia. A general description of the genus of lyssaviruses and the family of coronaviruses is given. The potential for virus carriage in relation to lyssaviruses and coronaviruses in bat populations of two geographically distant regions is considered.
415-428
ORIGINAL RESEARCHES
A preregistered meta-meta-analysis on the global distribution of Hepatotropic Viruses

Abstract
Introduction. Hepatotropic viruses (HAV, HBV, HCV, HDV, and HEV) significantly impact global health, with varying prevalence across regions.
Objective. This study aims to systematically consolidate data from diverse meta-analyses to provide a contemporary reference on virus distribution and prevalence.
Materials and methods. Adhering to PRISMA guidelines, the study utilized a mixed effects model for data integration. Quality evaluation was carried out with QUOROM and AMSTAR tools, with heterogeneity assessed via the Higgins I2 statistic, Q-statistic and Tau squared (τ2) values.
Results. The study analyzed 86 meta-analyses from 56 studies (2017–2022) with minimal overlap. Prevalence rates by region were as follows: MENA – 29.2%, Afghanistan – 9.14%, Africa – 8.10%. Prevalence rates by virus type: HAV – 82.5%, HBV – 8.6%, HCV – 15.1%, HDV – 8.9%, HEV – 13.9%, dual HBV-HCV coinfection – 2.2%. Prevalence rates by risk groups: general population – 8.3%, healthcare workers – 4.0%. Continent-specific HBV-HCV prevalence rates: Africa – 9.2%, China – 6.9%, others. HCVprevalence rates among at-risk groups: healthcare workers – 5.58%, hemodialysis patients – 34.8%. Regional HCV rates: Africa – 7.42%, Middle East – 25.30%.
Conclusion. Diverse global hepatotropic virus prevalence patterns are influenced by multifaceted factors. MENA faces higher rates due to healthcare challenges, while Africa struggles with limited resources. Tailored public health strategies, including vaccination and awareness campaigns, are essential to alleviate burdens and enhance global health. This consolidated data serves as a valuable resource for informed decision-making.
429-440
Evaluation of anti-HIV-1 (Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodeficiency virus type 1) activity of 6HP and 3TC in vitro using MT-4 cell line variants with different replicative activity
Abstract
Introduction. Chemotherapy of HIV infection remains the only means of treating the disease. The process of development new and improving previously developed drugs is therefore considered a priority. One of the preclinical stage of drug efficacy testing is research in the virus-cell model system in vitro.
The aim. To evaluate the antiviral efficacy of nucleoside reverse transcriptase inhibitors (NRTIs) 6HP and 3TC during HIV-1 replication in the neoplastic MT-4 cell line.
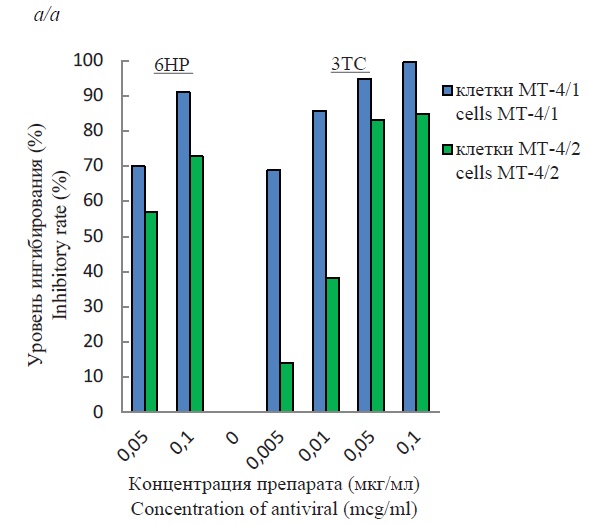
Materials and methods. Two variants of the CD4+ T-lymphocyte MT-4 cell line (MT-4/1 and MT-4/2) transformed by Human T-lymphotropic virus type 1 (Retroviridae: Orthoretrovirinae: Deltaretrovirus: HTLV-1), with different levels of HIV-1 replication were used. Drugs ammonium-3’-azido-3’-deoxythymidine-5’-carbomoylphosphonat (6HP) and 2’,3’-dideoxy-3’-thiacytidine (3TC) were used to suppress the virus.
Results and discussion. The replication activity of HIV-1 was observed to be higher in the MT-4/2 line than in the MT-4/1 line for different strains of the virus. The use of each of the substances separately showed a more significant inhibition of viral activity in MT-4/1 than in MT-4/2 cells. When used together, the inhibition level was almost the same in all cases and ranged from 87‒96% for the MT-4/1 line and 83‒89% for the MT-4/2 line. High efficacy was observed when using lower concentrations of drugs compared to individual use.
Conclusion. The combined use of NRTIs 6НР and 3TС is promising for the treatment of HIV-infected patients at different stages of infection and with different levels of viral load.
441-448
Mutations in human cytymegalovirus (Orthoherpesviridae: Herpesvirales: Cytomegalovirus: Cytomegalovirus humanbeta 5) UL97 gene lead to increase in viremia duration and poor antiviral response in recipients of allogeneic hematopoietic stem cells
Abstract
Introduction. Human cytomegalovirus (Orthoherpesviridae: Herpesvirales: Cytomegalovirus: Cytomegalovirus humanbeta 5) (HCMV) is one of the most commonly detected viruses in recipients of allogeneic hematopoietic stem cell (allo-HSCT) transplants. However, the emergence of resistance to antiviral drugs such as ganciclovir (GCV) poses a challenge in managing these patients.
This study aims to investigate the prevalence and impact of mutations in the HCMV UL97 gene associated with resistance to GCV on the course of infection among allo-HSCT patients.
Materials and methods. The study examined the association between UL97 mutations and the clinical course of HCMV infection in allo-HSCT patients. Genetic sequencing was performed to identify mutations, and their impact on viral replication and resistance to GCV was assessed.
Results and discussion. Six mutations were identified (D490A, T502A, C592G, C592F, E596G, C603W). C592G, C592F, E596G, and C603W are associated with resistance to antiviral drugs, while D490A and T502A described for the first time. When comparing patients with wild-type and those carrying the mutant variant, several parameters of peripheral blood were significantly lower in the former group. The median time to peak viral load following allo-HSCT, duration of viremia, and rate of virological response to high-dose therapy also differed significantly between the two groups.
Conclusion. It was shown that approximately one third (4 out of 14) of allogeneic stem cell transplant recipients had mutations associated with resistance to GCV. Patients carrying the mutant variant of HCMV had longer viremia and took longer to achieve a negative virological test result after starting high-dose therapy. Performing genotyping may help make more evidence-based therapeutic decisions.
449-458
Evaluation of the effectiveness of chemical inactivation and immunogenicity of the Omicron variant of the SARS-CoV-2 virus
Abstract
Introduction. The rapid spread of coronavirus infection COVID-19 among the population of many countries around the world has contributed to the emergence of many genetic variants of SARS-CoV-2. Compared to previous coronavirus variants, the new Omicron variants have shown a noticeable degree of mutation. Virus inactivation is one of the most important steps in the development of inactivated vaccines. The chemical inactivation agents currently used are β-propiolactone and formaldehyde, but there is no uniform standard for designing and specifying the inactivation process.
Objective. Evaluation and comparison of the effectiveness of chemical inactivation of two agents, formaldehyde and β-propiolactone against immunogenicity of the Omicron variant of the SARS-CoV-2 virus.
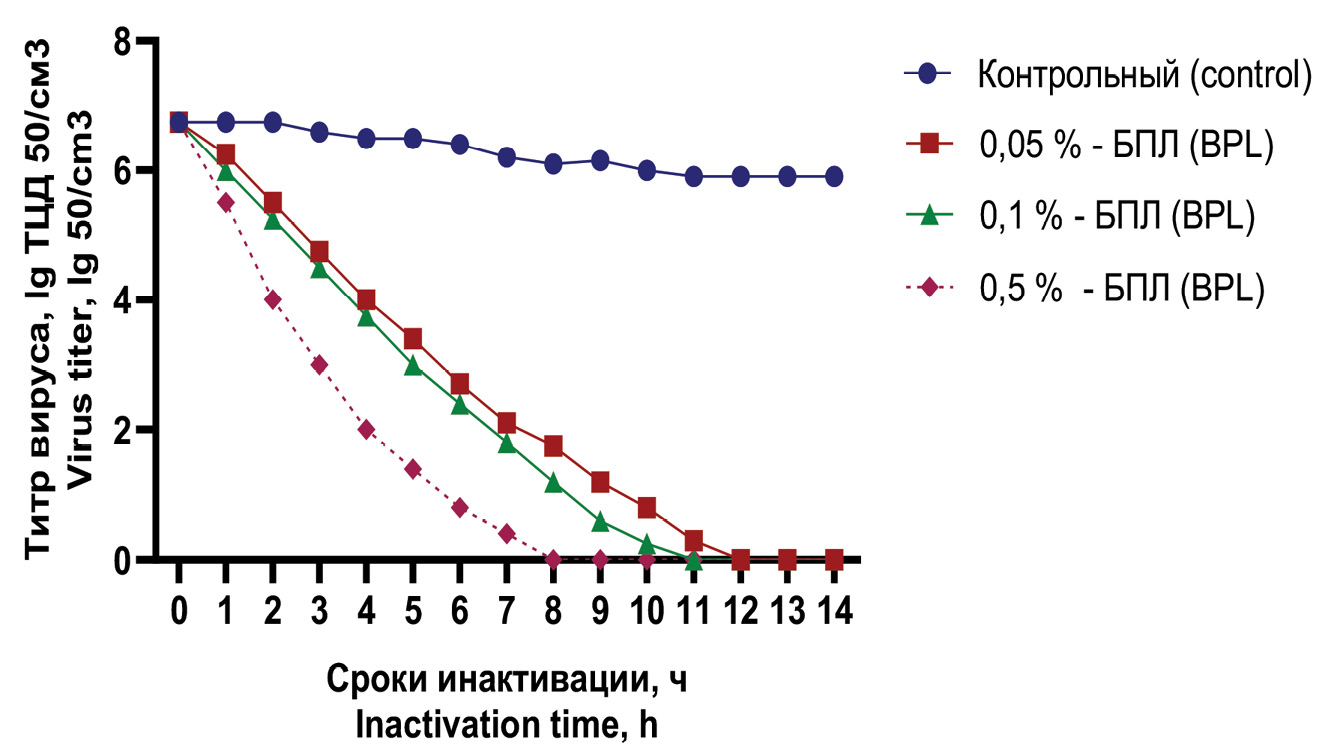
Materials and methods. Nasopharyngeal swabs were used to obtain the SARS-CoV-2 Omicron variant virus. Vero cell culture was used to isolate, reproduce, titrate the virus, and perform a neutralization reaction. The kinetics of studying the inactivation of the virus by chemical agents such as formaldehyde and β-propiolactone was carried out.
Results. Studies have been conducted to comparatively evaluate the effectiveness of chemical agents used to inactivate the SARS-CoV-2 virus of the Omicron variant, planned for use in the production of an inactivated whole-virion vaccine. Formaldehyde and β-propiolactone were used as inactivation agents in concentrations of 0.05, 0.1, 0.5% of the total volume of the virus suspension. It has been established that complete inactivation of the virus by formaldehyde in the concentrations used at a temperature of 37 °C occurs within up to 2 hours, and when using beta-propiolactone, within up to 12 hours.
Conclusion. Inactivated virus samples have different antigenic activity depending on the concentration of the inactivation agents used. The most pronounced antigenic activity is manifested in samples of the pathogen that were treated with an inactivation agent at a mild concentration of 0.05%. Increasing the concentration of inactivation agent by 5 or more times leads to a significant decrease in the antigenicity of the SARS-CoV-2 virus. With the inactivation modes used, the loss of biological activity of the virus occurs faster and antigenicity is retained largely when treated with formaldehyde.
459-469
Variability of non-structural proteins of HIV-1 sub-subtype A6 (Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodeficiency virus-1, sub-subtype A6) variants circulating in different regions of the Russian Federation
Abstract
Introduction. HIV-1 non-structural proteins are promising targets for vaccine development and for creating approaches to personalized medicine. HIV-1 sub-subtype A6 has become the dominating strain in Russia. However, the geographic, economic and demographic characteristics of the country can contribute to the formation of differences between A6 variants circulating in different regions.
The aim of the study is a comparative analysis of the consensus sequences of non-structural proteins in A6 variants circulating in the Amur Region, in Arkhangelsk, Irkutsk and Murmansk.
Materials and methods. 48 whole blood samples obtained from HIV-infected patients without experience of therapy observed at the AIDS Centers in Arkhangelsk, Irkutsk, Murmansk and Amur Region were analyzed. HIV-1 whole-genome nucleotide sequences were obtained and were subtyped. Consensus sequences of sub-subtype A6 variants non-structural proteins for each analyzed region were formed. Furthermore, reference sequences of sub-subtype A6 non-structural proteins were formed based on whole-genome sequences retrieved from the international Los Alamos database. Comparison of consensus sequences and references was performed using the MEGA v.10.2.2 and the PSIPRED programs.
Results. Vif, Vpr and Nef reference sequences have been obtained for HIV-1 sub-subtype A6. There was not difference in consensus sequences of Vpr in different regions. Characteristic features were found for consensus sequences of Tat, Rev, Vpu, Vif and Nef proteins in different regions.
Conclusion. A limitation of the study is a small sample size. Overall, the results demonstrate the existing diversity of non-structural proteins in sub-subtype A6 variants in different regions and indicate the relevance of studying the polymorphism of non-structural proteins of virus variants in different regions.
470-480
Comparative analysis of whole-genome sequences of African swine fever virus (Asfarviridae: Asfivirus) isolates сollected on the territory of the left bank of the Dnieper River in 2023
Abstract
Introduction. The lack of data on the whole-genome sequences of African swine fever virus (ASFV) variants circulating on the territory of the left bank of the Dnieper River complicates the understanding of the molecular evolution of the virus and the character of the epidemic process development in Russia and Ukraine. Understanding the genetic divergence and phylogenetic relatedness of isolates can largely adjust the strategy of general and specific prevention of the disease.
The aim of the study – search and description of unique mutations (deletions/insertions/substitutions) in isolates collected from domestic pigs in Donetsk, Luhansk and Zaporozhye regions in 2023; determination of relatedness and level of homology with reference strains of ASFV genotype II; sub-genotyping and clustering of isolates based on whole-genome analysis.
Materials and methods. The samples used were a culture suspension of porcine bone marrow (PBM) cells containing ASFV isolates obtained from pathologic material from domestic pig carcasses. Genomic DNA was prepared by purification and concentration of virus followed by phenol-chloroform extraction of total nucleic acid. The high-throughput sequencing process was performed using MGI technology. Consensus sequences were assembled by mapping reads to the reference genome of strain Georgia 2007/1.
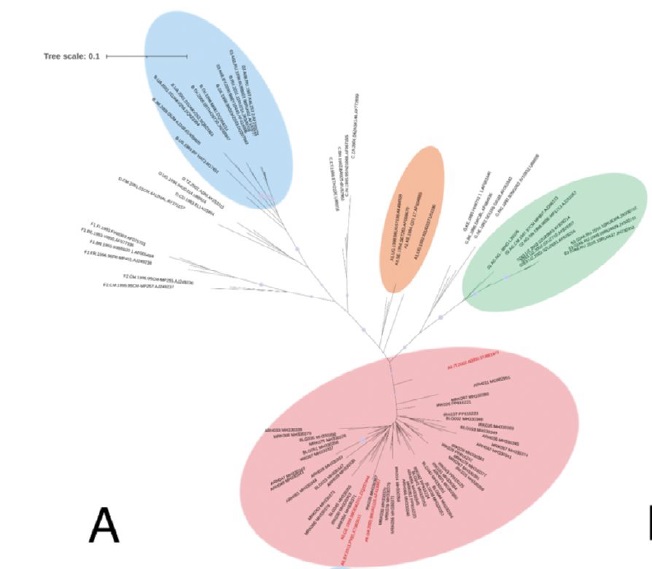
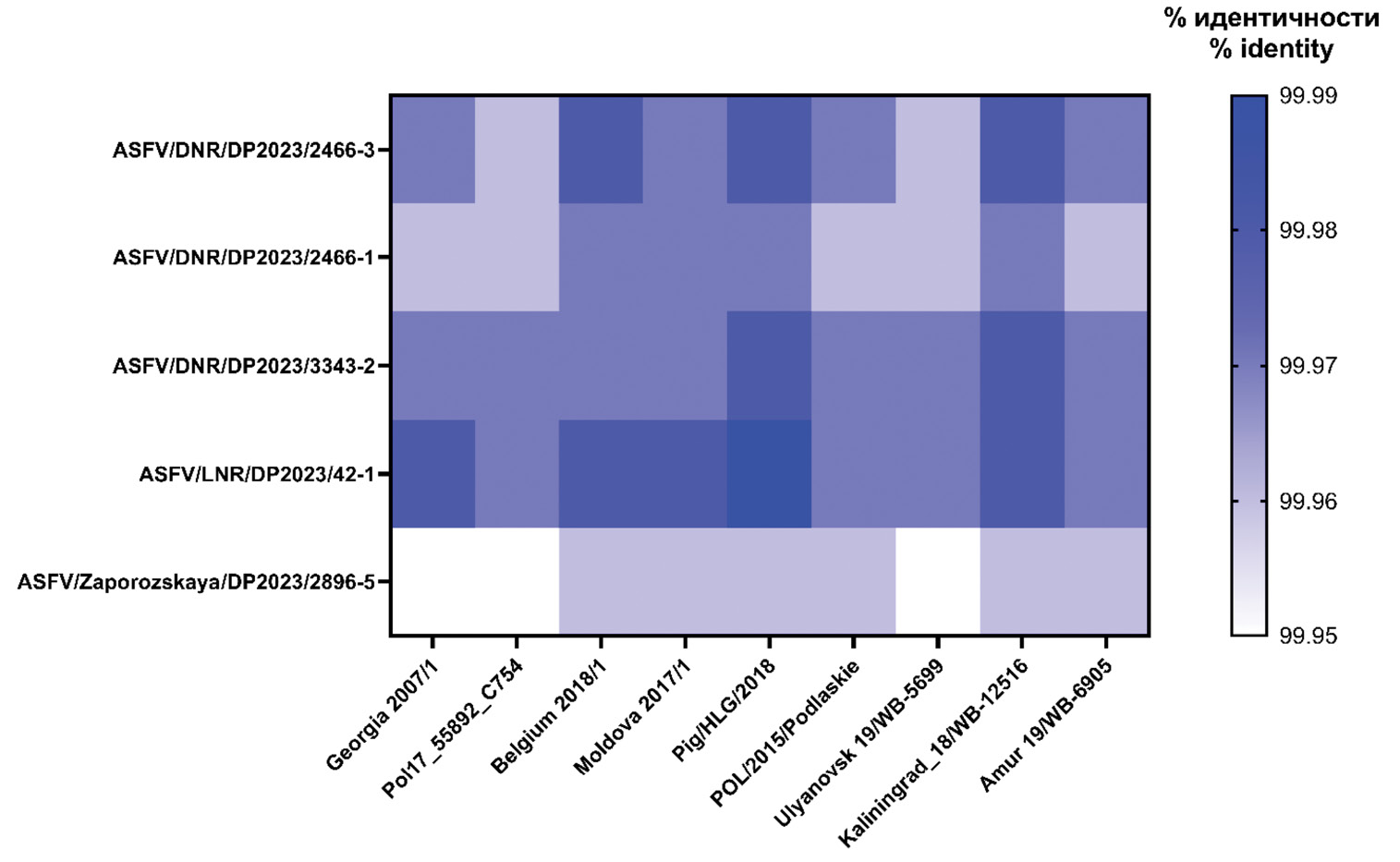
Results. All isolates are assigned to genotype II, have a monophyletic origin, are phylogenetically close to the clusters «Europe» (4/5) and «Bryansk 2021» (1/5), and are divergent from the original parental genetic variants that make up the enlarged clades. In addition, numerous substitutions in the loci of the multigene family MGF 110, 505, and 360, encoding virulence proteins, were detected in 4 isolates from Donetsk and Zaporozhye regions.
Conclusion. The phylogeny of the genotype II ASFV, which originated from the reference strain Georgia 2007/1, is shown to be sufficient for isolate differentiation. The presented data are of theoretical and practical importance for domestic and international ASFV surveillance.
481-494
Регистрационный номер и дата принятия решения о регистрации СМИ: серия ПИ № ФС77-77676 от 29.01.2020.