



Defective HIV proviruses: possible involvement in the HIV infection pathogenesis
- Authors: Bobkova M.R.1
-
Affiliations:
- I. Mechnikov Research Institute for Vaccines and Sera
- Issue: Vol 69, No 5 (2024)
- Pages: 399-414
- Section: REVIEWS
- URL: https://virusjour.crie.ru/jour/article/view/16678
- DOI: https://doi.org/10.36233/0507-4088-261
- EDN: https://elibrary.ru/pselci
- ID: 16678

Cite item

Abstract
This review article analyzes information obtained from a literature search on defective HIV genomes (HIV-1, Human Immunodeficiency Virus, Lentivirus, Orthoretrovirinae, Retroviridae). It discusses the origins of defective HIV genomes, their potential for transcription and translation, and the role of defective RNA and proteins in stimulating both innate and adaptive immunity. The article also explores their contribution to HIV pathogenesis, immune system hyperactivation despite successful antiretroviral therapy (ART), and the evolutionary processes in HIV proviral populations under ART. Additionally, it addresses challenges in reservoir elimination and HIV eradication that arise from the existence of defective HIV viruses.
Full Text
Introduction
Widespread implementation and improvement of antiretroviral therapy (ART) has significantly reduced morbidity and mortality of people living with HIV (PLHIV). The effect of ART is aimed at simultaneous restriction of several stages of viral replication and prevention of infection of new target cells, but complete cure of HIV infection remains unattainable due to the existence of reservoirs – both cellular (latently infected cells containing HIV proviral DNA) and anatomical (organs and tissues where viral replication continues) [1, 2].
For this reason, withdrawal of treatment even after a prolonged absence of viral load (VL) inevitably leads to rapid recovery of virus production. Lifelong treatment increases the life expectancy of PLHIV, almost approaching the average life expectancy for uninfected people, but can be complicated by the toxicity of ART drugs, drug interactions, treatment adherence problems, stigmatization, and significant economic costs.
The success of ART in clinical settings is usually assessed by the reduction of VL to an undetectable level (usually less than 20–50 copies of RNA/mL of blood) and an increase in absolute and relative CD4+ T-cell counts. Despite improved immune system function with successful ART, PLHIV have a higher risk of new diseases and death from serious non-AIDS-related complications compared to uninfected individuals of the same age. Among the high-risk conditions are non-AIDS-related cancers, chronic cardiovascular disease, liver disease and kidney disease. As currently established [3–5], this increased risk is due to the well-known phenomenon of increased markers of chronic inflammation and immune activation (hyperactivation) [2, 6–9]. Activation markers are independent of the effectiveness of VL suppression and continue to increase throughout life even in the most successful patients with long-term undetectable VL; the volume of reservoirs is also virtually independent of the success of ART [10, 11].
To date, the cause of HIV-associated chronic immune system activation in PLHIV remains incompletely understood. Several processes are thought to play a role in this phenomenon; these include microbial translocation [12], chronic coinfection with other pathogens such as cytomegalovirus [13], host genetics [14], lifestyle factors (e.g., smoking and drug use) [15] and the phenomenon of so-called low level viremia of HIV, the source of which is likely to be periodic reservoir activation [16–18].
Meanwhile, a significant number of studies in recent years have been devoted to the so-called «defective» (hereinafter without quotation marks) virus genomes (these include influenza virus, bunyaviruses, flaviviruses, alphaviruses, coronaviruses, picornaviruses) [19, 20], including HIV proviruses (defective provirus). The very fact of their existence was described a long time ago – in 1997, a significant presence of HIV proviral genomes with deletions of different sizes was found in peripheral blood mononuclear cells of PLHIV [21]. As it became evident later, defective proviruses are not just common, but constitute 90% [11] to 99.8% [10] of the entire proviral population.
A common feature of defective proviruses is their inability to produce infectious viral particles, and for a number of years the consensus view was that they were a «dead-end product» of HIV replication, forming a «graveyard» of viruses [22]. Recent data provide evidence to completely revise this view and argue that HIV proviruses in patients on ART who have achieved complete suppression of HIV replication (hereafter referred to as ART patients) are not defective in the general sense, but rather represent incomplete forms of proviruses capable of producing cell-associated RNA transcripts and expressing novel HIV-related proteins. This information provides a basis for analyzing the possible involvement of defective viruses in the pathophysiology of HIV infection in the setting of successful ART, including the phenomenon of chronic immune system activation. Some of what is currently known about defective HIV proviruses will be summarized in this review.
Origin of defective HIV genomes
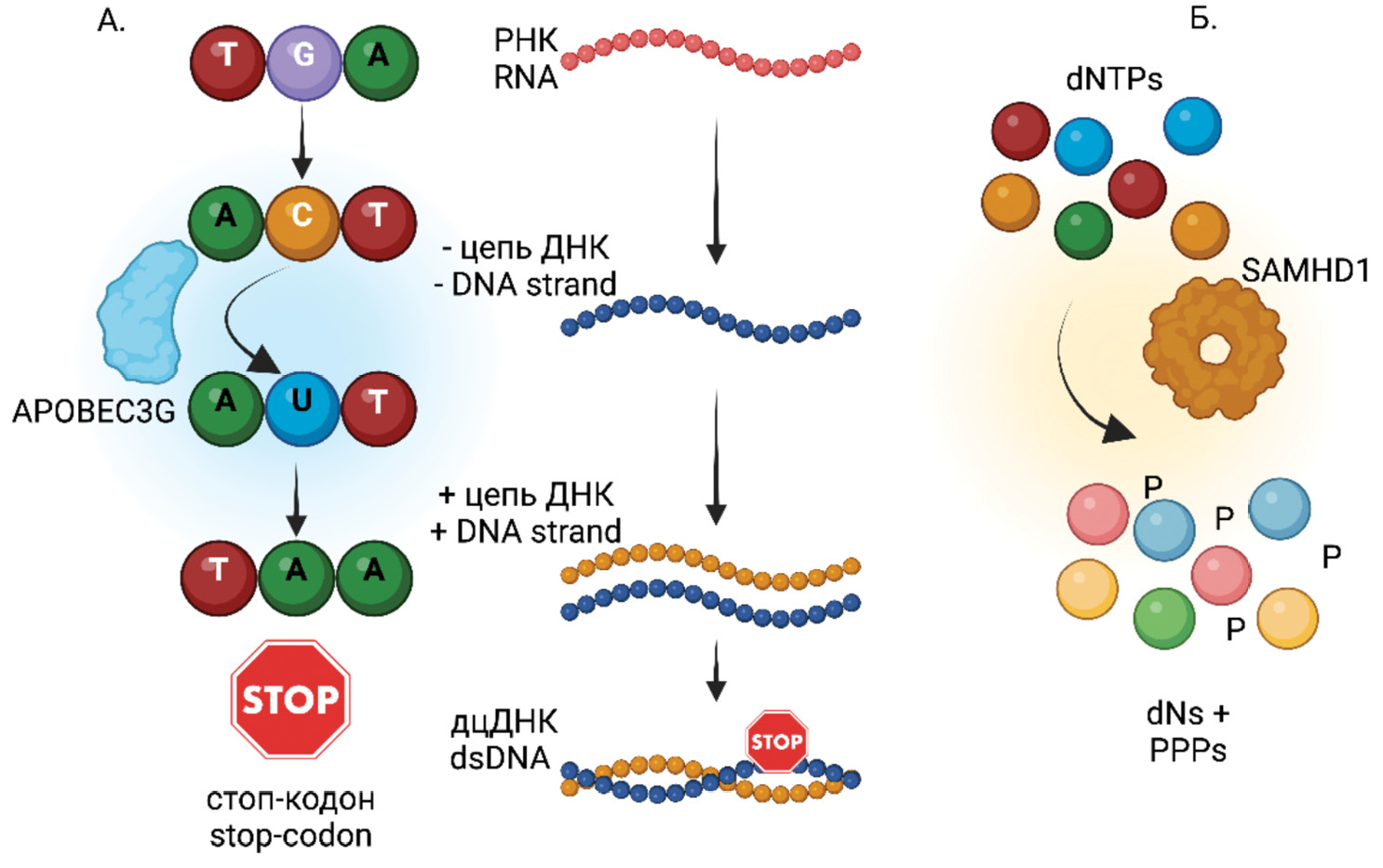
The formation of defective HIV genomes is the result of a combination of errors occurring at different stages of viral replication, the action of host restriction factors, and subsequent selection under the control of the immune system [23]. The most vulnerable to errors is undoubtedly the stage of reverse transcription. HIV reverse transcriptase (RT) works with low accuracy at the stage of cDNA synthesis and makes about 10−5 erroneous nucleotide insertions (mutations)/nucleotide/replication cycle, and the enzyme has no proofreading activity. In addition, mutations and deletions are generated at the moment of template switching during reverse transcription, when RT makes characteristic «jumps» [24]. Moreover, the reverse transcription process involves RT dissociation and re-initiation of synthesis on the template of genomic RNA, which leads to the formation of mutant and shortened HIV cDNA intermediates [23, 25‒27] (Fig. 1). Small deletions (one/few nucleotides) are also capable of causing noticeable damage to proviral DNA, manifested by stop codon formation or frameshift [22, 25].

Fig. 1. Causes of Defective HIV Virus Formation.
a ‒ deletions in HIV genome fragments; b ‒ G→A hypermutations, illustrated with APOBEC3G; c ‒ insertions; d ‒ stop codons; e ‒ mechanical DNA damage
Рис. 1. Причины формирования дефектных ВИЧ.
а ‒ делеции фрагментов генома ВИЧ; б ‒ гипермутации G→A на примере APOBEC3G; в ‒ инсерция (вставка); г ‒ стоп-кодоны; д ‒ механическое повреждение ДНК.
In addition to large and small deletions, defective proviruses can have «shortages» of 5’- and 3’-end genome fragments and inversions of genome regions, which are formed mainly at the RT stage, as well as hypermutations and defects in donor-acceptor splice sites [10, 27–29].
The cellular restriction factor SAMHD1 [30–34], which reduces the intracellular concentration of nucleotides in resting CD4+ T cells and myeloid cells, thus limiting the efficiency and completion of reverse transcription, may contribute to the formation of deletions. Another host cell restriction factor, APOBEC3G, also acts during the reverse transcription step, acting as a cytidine deaminase (C→U) within the newly synthesized minus-strand of the cDNA. The result is a hypermutation of HIV caused by the replacement of guanine by adenine (G→A) within the plus-strand of DNA and associated with the formation of stop codons [32, 35, 36] (Fig. 2).

Fig. 2. Contribution of Host Cell Restriction Factors to the Formation of Mutations in the HIV Genome During Reverse Transcription.
a ‒ APOBEC3G – a cytidine deaminase; b ‒ SAMHD1 – a phosphohydrolase.
Рис. 2. Вклад рестрикционных факторов хозяйской клетки в формирование мутаций в геноме ВИЧ на этапе обратной транскрипции.
а – APOBEC3G – цитидиндезаминаза; б – SAMHD1 – фосфогидролаза.
Finally, errors in the provirus sequence can also appear at the stage of its integration into the chromosomal DNA of the cell. This complex process, described by many authors [30, 37, 38], will not be described in detail here; we will only recall that the last stage of integration, strand transfer, requires «completion» of provirus end fragments with the participation of host DNA repair enzymes. The mechanism of damaged DNA repair is known to be error-prone [39], which serves as the source of defective proviruses.
Defective genomes can be transcribed
Under conditions of successful ART, most cells carrying proviral DNA are in a latent state and do not produce viral particles; however, up to 7% of proviruses have been shown to remain transcriptionally active [40]. Transcription in latently infected cells occurs in the same way as in cells with active HIV replication and involves several steps described in detail in the literature [2, 41].
Briefly, the HIV long terminal repeat (LTR) acts as an enhancer and promoter that recruits host cell transcription activators, repressors, chromatin remodeling factors, and the RNA polymerase-RNAP II complex, each of which affects activation or repression of transcription. The HIV regulatory protein Tat binds the TAR (trans-activation response) loop, an element at the 5’-end of the HIV primary transcript, and recruits PTEFb (positive transcription elongation factor b), a cofactor that increases the processivity of RNAPII. Its activity results in full-length HIV RNA transcripts. Some of them subsequently become genomic RNA and are incorporated into new viral particles; unspliced messenger RNA (mRNA) is also required for the synthesis of Gag and Pol proteins, while another part of RNA undergoes splicing to form Env, Vif, Vpr and Vpu (partially (single) spliced mRNA) and Tat, Rev and Nef (multiple spliced mRNA variant) proteins (Fig. 3).

Fig. 3. Splicing Variants of Full-Length HIV RNA.
Рис. 3. Варианты сплайсинга полноразмерной РНК ВИЧ.
A significant proportion of proviral sequences found in PLHIV receiving ART are defective and contain mutations that may affect the LTR structure, major donor splicing sequences, and the ψ-packing element (a hairpin RNA sequence required for genome dimerization during packaging [42]). Transcripts of both intact and defective proviruses have been found to be detected in comparable amounts in the blood of patients with fully suppressed VL [11, 28, 43], with the transcriptional outcome of the latter depending on the nature and extent of the defect.
The absence of the promoter 5’-terminal fragment leads to the formation of non-canonical RNA transcripts containing env and nef sequences (Fig. 3); in this matrix case, alternative sites for transcription initiation are apparently used, for example, such sites are present in the env gene [28]. If the defect affects donor splicing sites, the problem is solved by using alternative splicing mechanisms [44]; intragenic cis-acting elements that interact with the transcriptional apparatus of the cell actively interfere with this process.
Another way to generate transcripts on the template of defective proviruses is the synthesis of antisense RNA on the complementary DNA strand starting from the 3’-LTR. The existence of such transcripts has been confirmed [45], but it is still unclear whether antisense transcription is regulated by the same signaling cascades as transcription from the 5’-LTR.
Often the tat/rev region of the genome is deleted in defective viruses; in these cases, the level of transcription without Tat protein is significantly reduced, and RNA export from the cell nucleus is impeded in the absence of Rev protein [10].
Finally, the presence of deletions and stop codons leads to premature arrest of mRNA synthesis, and such short transcripts are also found in ART patients [28].
Thus, even in the case of ART success, unspliced and partially spliced RNAs derived from intact proviruses, as well as a variety of non-canonical RNAs originating from defective proviruses, are detected at a significant level in patient cells.
Defective genomes can produce proteins
The presence of a significant amount of unusual RNAs in latently infected cells has naturally raised the question of whether they are capable of serving as a template for protein synthesis. The answer to this question has already been obtained, and it is now well known that a significant fraction of non-canonical RNAs provides translation of proteins as abnormal as their nucleotide templates [17, 28, 44].
Most often, provirus defects – deletions, inversions, and point mutations - are associated with disrupted ORFs, and the obvious way out of this situation is to use alternative ORFs; such, usually shortened (cryptic), proteins can indeed be found in successful ART patients [22, 28, 46]. Alternative RNA splicing similarly results in aberrant proteins [44].
Cases of alternative translational start codons (e.g., instead of the traditional AUG codon, protein synthesis initiation can occur with the participation of the CUG codon), translation of antisense RNAs, as well as the mechanism of leaky ribosomal scanning, when the ribosome, having encountered an «unfavorable nucleotide context» around the start codon, continues to move along the RNA molecule in search of an optimal binding site, have also been described [23].
The question of whether abnormal proteins can form virus-like particles is still open. No less interesting is the question of their immunogenicity, and in this respect some certainty has already been achieved. It has been shown, for example, that defective ribosomal products (DRiPs) are rapidly degraded via the proteasomal pathway and “loaded” onto MHC-I molecules that exhibit them on the cell surface [28]. The same fate befalls proteins – products of antisense RNAs, and they cause the formation of circulating antibodies [47]. Finally, non-canonical HIV peptides activate CD8+ T cells from PLHIV receiving ART, which also indicates their immunostimulatory activity [48].
All these observations suggest that defective HIV proviruses, which are unable to produce viable progeny and are therefore not fully «alive» (one researcher called them «zombie viruses» [22]), retain the ability to generate macromolecules important for the pathogenesis of infection. The question of how cryptic transcripts and proteins – products of defective proviruses – become immunomodulators and influence the host immune responses is being actively studied.
Non-canonical RNAs and natural immunity
Several types of viral nucleic acids participate in the HIV reproduction cycle: viral RNA, cDNA, and RNA transcripts of proviral DNA in the cytosol, and proviral DNA and RNA transcripts in the cell nucleus. All these molecules can act as intracellular pathogen-associated molecular patterns (PAMPs) and interact with pathogen recognition receptors (PRRs) [23].
PRRs that recognize DNA molecules primarily include the cytosolic DNA sensor cGAS (cyclic guanosine adenosine synthase) [49]. Normally, DNA should not be present in the cytoplasm, and detection of foreign DNA of viral or bacterial origin is a crucial element of immunity in many organisms. In mammalian cells, the cGAS-STING (cyclic GMP-AMP synthase – stimulator of interferon genes) system is available for this task.
After DNA binding, cGAS acts as an allosteric trigger of the reaction between GMP and AMP to form cyclic cGAMP, which, in turn, binds molecules of stimulator of interferon genes (STING) that stimulates phosphorylation of IRF3 (interferon regulatory factor 3). The chain of events is completed by the movement of IRF3 into the cell nucleus and transcription of interferon α and ß genes (Fig. 4) [49].

Fig. 4. HIV RNA and DNA Molecules as Stimulators of Innate Immunity.
DNA sensors: IFI16 and the cGAS-STING complex; RNA sensors: MAVS and RIG-1; IRF3 – interferon regulatory factor.
Рис. 4. Молекулы РНК и ДНК ВИЧ – стимуляторы естественного иммунитета.
ДНК-сенсоры: IFI16 и комплекс cGAS-STING; РНК-сенсоры: MAVS и RIG-1; IRF3 – фактор регуляции интерферона.
The IFI16 (gamma-interferon-inducible protein 16) protein is another DNA sensor that also recruits STING after binding to DNA. Subsequent events lead to the induction of interferons and pro-inflammatory cytokines. In addition, CD4+ T-cell pyroptosis, an accelerated programmed cell death accompanied by active cellular release of interleukin (IL) 1β and IL-18, is observed [50].
HIV RNA, in turn, attracts the attention of cytosolic RNA sensors; one example is the tandem of MAVS (mitochondrial antiviral-signaling protein) and RIG-1 (retinoic acid-inducible gene I) proteins [51].
MAVS protein is located on the outer membrane of mitochondria, peroxisomes and endoplasmic reticulum. During viral infection, the cytosolic protein RIG-1 (sometimes replaced by the Toll-like receptor TLR-3) detects the presence of virus and binds to MAVS. After the attachment of several cellular proteins [51], a MAVS-signaling complex is formed, which results in the phosphorylation and movement of the already mentioned IRF3 factor into the nucleus, followed by the traditional activation of transcription of interferon and proinflammatory cytokine genes (Fig. 4) [23].
Thus, the infected cell always contains HIV-associated DNA and RNA molecules that act as PAMPs and initiate the inflammatory response of the natural mechanisms of the immune system. The data directly indicating the participation of non-canonical DNA and RNA – products of defective HIV genomes – in these processes are not yet available in the literature. However, since the described process of RNA/DNA recognition is nonspecific, there is no reason to believe that these molecules can behave fundamentally differently from intact RNA, and at present most experts are convinced that it is defective RNA and DNA that are the main triggers of the signaling cascades of the inflammatory response of the natural immune system in patients in the absence of HIV infection.
Protein products of defective HIV genomes and immune system hyperactivation
Non-canonical RNAs formed during transcription of defective HIV proviruses also have various defects; therefore, as mentioned above, if such RNAs are destined to become a template for translation, the resulting (aberrant, cryptic) proteins differ from normal virus proteins in size, structure, and antigenic properties [28, 44]. Nevertheless, the defects do not necessarily affect all genes of the virus, and if the reading frames and integrity of individual genes are preserved after the completion of RNA maturation processes (splicing and modification), such RNAs may well become a template for translation of full-fledged HIV proteins. Thus, [46] demonstrated the prolonged presence of Nef and Gag proteins in ART patients after reaching undetectable VL. The authors of the study [52] observed similar results and made valid conclusions that Nef protein production in the absence of active viral replication is at least partially explained by the presence of a pool of cells carrying translation-competent defective proviruses.
Aberrant proteins along with intact HIV proteins retained the ability to induce CD4 and CD8 T-cell response [22, 28]. In [4], the intensity of bands on Western blot was used as a surrogate marker of the amount of viral proteins in the blood plasma of ART patients, while estimating the amount of intact proviruses in parallel. As it turned out, antibodies to HIV proteins persisted in such patients for 10–20 years after reaching undetectable VL, i.e. cessation of virus replication. By this time, there were almost no intact genomes left, and the pool of proviruses was represented mainly by defective genomes, which served as a template for the synthesis of viral protein antigens.
Thus, the set of proteins in the cell carrying the defective provirus includes intact and cryptic HIV proteins, and are capable of inducing an immune response, humoral and cellular (Fig. 5). Regardless of the direction of this response, immune cells in ART-successful patients are involved in a continuous process of antigenic stimulation, which, according to many experts, is the cause of chronic activation of the immune system [4, 17, 22, 28]. The same phenomenon underlies the so-called «exhaustion» of HIV-specific CD8+ cytotoxic lymphocytes [53, 54].

Fig. 5. Formation of the Cellular Immune Response to Intact and Defective HIV Proteins.
Рис. 5. Формирование клеточного иммунного ответа на интактные и дефектные белки ВИЧ.
The search for direct evidence of an association between the persistence of incomplete (i.e., defective) cell associated HIV RNA, the humoral response to HIV (antibody concentrations), and inflammatory markers (cytokine IL-6, D-dimer) led to the discovery of a direct correlation between them [4]. Similarly, in [43], the content of short (i.e., non-canonical) cell associated RNA was strongly associated with the degree of chronic immune activation as determined by co-expression of HLA-DR and CD38 on CD8+ T cells. These and other observations provided evidence for a link between defective HIV expression and immune activation; the authors note that this phenomenon, in turn, may lead to a lack of CD4+ cell counts despite high ART efficacy (i.e., in the absence of viral replication); clinicians refer to such findings as discordant.
Taken together, the results of studies of defective HIV RNAs and proteins bring us ever closer to the conclusion that persistent defective proviruses, especially those that are transcriptionally active, are not «genetic garbage» irrelevant to HIV pathogenesis and treatment, but rather, during successful ART, provide a continuous (and unfortunately redundant) interaction between «residual expression» of HIV genes and the immune system [10, 55].
Defective HIV genomes and methods of reservoir assessment
The persistence of latent viral genomes in PLHIV receiving ART is a major obstacle to HIV cure [1, 2]. Viral reservoirs are primarily cells containing HIV genomes that have the ability to produce infectious virions. Developments aimed at eradication or functional cure of HIV infection must necessarily be based on one or another method of quantifying the reservoir volume, allowing to measure the effect of the tested drug. It is important to emphasize here that a true reservoir can be considered only those cells in which latent virus can be induced (activated) with subsequent formation of new virions.
Accurate quantification of HIV cellular reservoirs is challenging and is based on two main approaches, either culture-based or molecular methods. The first group of methods (expensive and labor-intensive) does measure the number of inducible (“live” or replication competent) proviruses only, but the conditions of induction in cell culture differ significantly from natural conditions, and a significant proportion of proviruses cannot be induced. For this reason, the results of such methods appear to be many times underestimated in relation to the real volume of the reservoir. The second group, based on PCR variants, is simple and cost-effective, but these methods similarly overestimate the volume of reservoirs because they are unable to distinguish between intact and defective HIV genomes [29, 56]. Thus, a single simple approach that would allow simultaneous assessment of genetic integrity and reservoir inducibility is still lacking. Attempts to combine the merits of each approach into a single method are constantly being made, and many of them demonstrate impressive results [2], but they still do not allow comparison of data obtained in different laboratories around the world. Nevertheless, it is possible to make comparative evaluations in the dynamics of patient follow-up or between groups of patients studied in the same experiment. This is how the data concerning the study of the composition of the population of persistent HIV genomes (proviral landscape) were obtained.
Clonal expansion and activation of latent HIV proviruses
The origin of the so-called “residual viremia”, i.e. the presence of single copies of HIV RNA in the plasma of patients with VL not detected by conventional methods, has been debated in the literature for a long time [18, 57–59] and will not be discussed in detail in this review. Briefly, the generally accepted view is that the appearance of new viral particles in such patients is not mainly due to ongoing viral replication, which is inhibited by ART drugs, but is the result of virus production from latently infected cells that occasionally proliferate during clonal expansion (Fig. 6).

Fig. 6. Origin of HIV Viral Particles.
a ‒ ongoing HIV replication with repeated infection cycles; b ‒ clonal expansion followed by periodic activation of the provirus.
Рис. 6. Происхождение вирусных частиц ВИЧ.
а – продолжающаяся репликация ВИЧ с повторными циклами заражения; б – клональная экспансия с последующей периодической активацией провируса.
Clonal expansion is the process of rapid cell division resulting in the multiplication of genetically identical clones of cells from a single parent cell. Evidence of such a development in latently HIV-infected cells was obtained in studies analyzing the integration sites of proviral DNA in viruses isolated from ART patients with the absence of VL.
The main hypothesis of these studies was based on the fact that the incorporation of provirus into chromosomal DNA is a random process, therefore, in case of repeated cycles of infection, different sites of provirus localization in host DNA will be detected. If the source of the virus is the same resting cells containing the same type of DNA with the same integration point, genetic analysis will reveal complete uniformity of proviruses and their localization, since independent integration of HIV DNA with identical localization in different cells is obviously impossible (Fig. 7). It is the latter variant that was found in the mentioned above studies [60, 61]. It is important to note that for the majority of latent cells, division during expansion is not a reason for provirus activation, and at the end of mitosis they retain their «inactive» state.

Fig. 7. Variability of HIV and Provirus Integration Sites Under Conditions of ongoing replication (a); clonal expansion (b).
Рис. 7. Вариабельность ВИЧ и участков интеграции провируса в условиях продолжающейся репликации (а) и клональной экспансии (б).
Understanding the reasons for provirus activation in resting T cells is more complicated, and there appear to be several possible reasons. The most discussed topic is the influence of the integration site of the proviral genome into chromosomal DNA on the probability of its transcription. A growing body of evidence suggests that integration of HIV provirus near transcriptionally active sites on the chromosome (provirus genomic context) may influence its expression (Fig. 8) [17, 55, 62]. A provirus embedded near an actively transcribed host gene has a better chance of its own expression because it presumably finds itself in a favorable environment of epigenetic factors promoting transcription. One of such factors turned out to be H3K36me3, a modified histone H3 known for its participation in human gene expression [63]. Other causes of HIV provirus activation include the impact of physical and chemical factors, infectious diseases, vaccination and the level of hyperactivation of the immune system as a whole [18, 64].

Fig. 8. Effect of the Genomic Context of the HIV Provirus on Transcription Efficiency.
a ‒ integration into a euchromatic region; b ‒ integration into a heterochromatic region of the human genome.
Рис. 8. Влияние геномного контекста провируса ВИЧ на эффективность его транскрипции.
а – интеграция в участок эухроматина; б – интеграция в гетерохроматиновую область генома.
These considerations apply primarily to intact proviruses, and there are almost no special studies evaluating the transcription efficiency of defective HIV genomes. The answer to this question requires technically challenging experiments because it requires a combined assessment of provirus integrity and transcriptional activity in the same cell. In a recent unique study, a single cell fluorescence in situ hybridization (FISH) method was used to identify HIV-infected cells that express viral RNA during ART [65]. As shown, almost all HIV proviruses sequenced from cells containing cell-associated HIV RNA were defective. Even assuming that the transcriptional efficiency of such proviruses is reduced relative to intact genomes, the significant prevalence of defective HIV variants should not be overlooked; even low levels of transcription and translation of these persistent proviruses may be sufficient to induce and maintain inflammatory and immune responses in ART patients [23, 28].
Evolution of the population of latent HIV proviruses
Thus, both types of proviruses – intact and defective – are involved in the maintenance of chronic activation of the immune system in ART patients, but, as it turned out, the proviral landscape undergoes changes in the dynamics of treatment.
First, analysis of intact proviral sequences in PLHIV before and after ART initiation indicates that the vast majority of the latent intact HIV reservoir in ART patients consists of viruses circulating at the time of treatment initiation. Once undetectable VL is reached, evolutionary mechanisms, primarily CD8+ cytotoxic cells, have nothing to work with and no appreciable evolutionary events in the viral population occur during ART [66]. These results again support the view that HIV replication is not a mechanism for maintaining the HIV reservoir during successful therapy, and once ART is initiated, periodic activation of reservoirs containing homogeneous proviral genomes becomes the main source of VL (Fig. 9).

Fig. 9. Key Events in the Evolution of the HIV Proviral Population.
a ‒ the period from infection to the start of ART; b‒d ‒ clonal expansion as the main source of HIV; c ‒ selection of proviruses integrated into heterochromatic regions of the host genome; d ‒ selection of defective proviruses. Stages C and D occur in parallel.
Рис. 9. Основные события эволюции популяции провирусов ВИЧ.
а – период от заражения до начала АРТ; б–г – клональная экспансия как основной источник вирусной нагрузки; в – отбор провирусов, встроенных в гетерохроматиновые участки генома хозяина; г – отбор дефектных провирусов. Этапы в и г происходят параллельно.
Second, the ratio of intact and defective viruses in the dynamics of ART appeared to be variable, and many studies have shown that the proportion of intact proviruses gradually decreases during treatment [10, 28, 55]. While before the start of ART it was up to 36%, 3 years after reaching undetectable VL the proportion decreased to 6% [22].
Third, important data on the localization of HIV proviral genomes were obtained. The results of a comparative study of the localization of intact and defective proviruses in elite controllers maintaining undetectable VL in the absence of ART look quite peculiar: as it turned out, the former are predominantly found in heterochromatin regions characterized by a low level of transcription, and the latter – in actively transcribed euchromatin regions [67]. This allowed the authors to hypothesize that the immune system forms a virus reservoir over time, favoring those proviruses that are located in «silent» regions of the host genome. Provirus reactivation experiments confirmed this assumption: relatively small groups of latently infected cells were easily induced with the formation of new virions, while the second, larger group of infected cells contained an intact provirus that was more resistant to reactivation [55]. The authors believe that intact proviruses integrated into heterochromatin regions and showing signs of «deep latency» appear to have advantages for selection and persist for a long time, probably due to very low or no proviral transcription and subsequent protection from antiviral immune recognition.
All of the above clearly indicates that the population of reservoir cells containing HIV proviruses undergoes significant evolution over time under the influence of the immune system, with the start of ART being a critical moment (Fig. 9). The overall picture is approximately as follows: HIV reservoir formation occurs within 7–10 days after infection, and the proviral landscape is quite diverse and includes defective proviruses. The main factor of the immune system influencing the process of proviral population evolution is the cytotoxic response of T-lymphocytes (CD8+) eliminating cells expressing foreign viral proteins [10].
The rapid evolutionary changes observed in the HIV population prior to ART are followed by stabilization and reduced provirus variability shortly after ART initiation. During treatment, the immune system continuously removes replicatively competent proviruses and selects intact proviruses that are in a state of «deep latency», i.e. as part of heterochromatin. Some authors [10, 19] call this process «natural cure» against the background of ART, which, however, never ends with virus eradication. In chronic HIV infection, cells carrying defective proviruses may not be recognized by the host as foreign due to inadequate presentation by the major histocompatibility complex [46]. This may explain the predominant clonal expansion and persistence of cells carrying defective proviruses, which over time take a dominant position [55].
In patients on long-term successful ART, more than 90% of proviruses are represented by defective genomes. These genomes strongly contribute to immune system hyperactivation and cytotoxic cell depletion, but are not a source of virus re-emergence when ART is discontinued. On the contrary, most intact proviruses are theoretically capable of producing replication-competent viruses, but in reality the proportion of such proviruses is small, and most of them cannot be brought out of latency by induction [68].
Defective proviruses and HIV eradication
The main practical goal of ART today is the indefinitely long-term maintenance of undetectable VL. For most PLHIV, this goal is achievable, but as treatment experience and new knowledge are gained, it is becoming clear that the absence of VL is not always a sufficient condition for restoring health, and the risks of comorbid diseases are increased even in the most successful ART patients. Complete eradication of HIV infection or its functional cure has become the new goal of researchers, and the literature of recent years has provided an avalanche of information on this topic.
Current strategies for curing HIV infection focus on either elimination of the HIV proviral reservoir, permanent inactivation of latent proviruses, or gene therapy approaches [1, 2]. Examples of the first group of proposed approaches include a variety of «kick-and-kill» technologies involving activation of the latent provirus pool followed by intensive ART and concomitant administration of a therapeutic vaccine. All activated proviruses are expected to produce viral particles that will be destroyed by CTL cells, and new rounds of infection will be prevented by ART inhibitors. An alternative «block-and-lock» approach from the second group of methods is based on the use of inactivating agents or specially designed transcriptional repressors that reliably and persistently inhibit the transcription of HIV proviruses, while the HIV genomes remaining in the chromosomal DNA will have to «peacefully coexist» with their gene environment, as endogenous retroviruses do. Finally, a large group of gene therapy methods is based on all existing technologies, such as CRISPR-Cas9 or zinc finger nucleases; the main problem here is the difficulty of delivering gene therapy tools to all infected cells of the adult organism.
The existence of defective proviruses may complicate the development and implementation of all of these methods, as it can be assumed that they will all have minimal impact on the presence of defective HIV genomes. Information on how transcription of defective proviral sequences is regulated is extremely limited, and it is unknown whether the latency reversing agents used by kick-and-kill to activate proviruses or the transcriptional repressors required for the block-and-lock strategy will affect the activity of intragenic cis-transcriptional elements and the expression of cryptic peptides [11, 55]. Furthermore, there is a high probability that defective proteins will not be recognized by a therapeutic vaccine [28] designed to target normal virus proteins.
Gene editing techniques depending on which sequences are targeted by engineered nucleases carry the risk of creating additional defective proviruses [28]. Furthermore, it has been reported that CRISPR-Cas9 can produce non-homologous DNA end joining and thereby promote the formation of transcriptionally active episomal elements [69].
The difficulties of creating universal methods for assessing the volume of HIV reservoirs have been mentioned above, and here we will only emphasize that in the absence of such methods it will be extremely difficult to assess the results of any of the approaches. This means that, while studying the ways to influence the latent HIV reservoir, it is necessary to develop studies aimed at a detailed understanding of the ways of formation, regulation and functional consequences of the activity of defective proviruses.
Conclusion
Observational and experimental work over the past decade has provided new insights into the events that occur in the HIV population after the establishment of undetectable VL levels as a result of ART. It has become clear that cessation of viral replication is not the «end of the story», and the pathogenesis of infection continues even in the face of prolonged HIV VL suppression. The main role in the «pathophysiology of successful ART» is played by reservoir cells containing HIV proviral DNA, and among them, paradoxically, are cells containing defective proviruses.
The traditional attitude to defective HIV genomes as an insignificant by-product of virus replication is being replaced by the belief that these proviruses can be biologically active, and the resulting RNA transcripts and proteins can serve as triggers of natural and adaptive immune response, followed by chronic activation of the immune system with long-term clinical consequences. It is the persistence of such proviruses that explains the long-term persistence of seropositivity and hyperactivation in ART patients.
Furthermore, defective proviruses may become a serious obstacle to the development of HIV eradication tools, which means that without special efforts aimed at eliminating cells carrying defective proviruses, none of the currently available strategies can be implemented.
The impact of defective genomes on the clinical outcome of infection and the evolution of the HIV population can be considered proven. Moreover, there has been rapid progress in understanding the molecular mechanisms that regulate the formation of defective HIV genomes and explain their controversial role in viral persistence. Finding a cure for HIV infection remains a challenge, but if successful, it will prove to be a critical step toward achieving an «AIDS-free generation». Research on defective proviruses should be key to further progress this field of study.
About the authors
Marina R. Bobkova
I. Mechnikov Research Institute for Vaccines and Sera
Author for correspondence.
Email: mrbobkova@mail.ru
ORCID iD: 0000-0001-5481-8957
D.Sci. (Biol.), Chief Researcher of the laboratory of biology of lentiviruses
Russian Federation, 105064, MoscowReferences
- Bobkova M.R. HIV infection cure strategies: basic methodological approaches and difficulties of their implementation. VICH-infektsiya i immunosupressii. 2020; 12(1): 22–31. https://doi.org/10.22328/2077-9828-2020-12-1-22-31 https://elibrary.ru/gsllxf (in Russian)
- Bobkova M.R. HIV Latency [Latentnost’ VICh]. Moscow: Chelovek; 2021. (in Russian)
- Grund B., Baker J.V., Deeks S.G., Wolfson J., Wentworth D., Cozzi-Lepri A., et al. Relevance of interleukin-6 and D-dimer for serious non-AIDS morbidity and death among HIV-positive adults on suppressive antiretroviral therapy. PLoS One. 2016; 11(5): e0155100. https://doi.org/10.1371/journal.pone.0155100
- Singh K., Natarajan V., Dewar R., Rupert A., Badralmaa Y., Zhai T., et al. Long-term persistence of transcriptionally active ‘defective’ HIV-1 proviruses: implications for persistent immune activation during antiretroviral therapy. AIDS. 2023; 37(14): 2119–30. https://doi.org/10.1097/qad.0000000000003667
- Trickey A., May M.T., Vehreschild J., Obel N., Gill M.J., Crane H., et al. Cause-specific mortality in HIV-positive patients who survived ten years after starting antiretroviral therapy. PLoS One. 2016; 11(8): e0160460. https://doi.org/10.1371/journal.pone.0160460
- Bandera A., Colella E., Rizzardini G., Gori A., Clerici M. Strategies to limit immune-activation in HIV patients. Expert Rev. Anti Infect. Ther. 2017; 15(1): 43–54. https://doi.org/10.1080/14787210.2017.1250624
- Elvstam O., Medstrand P., Jansson M., Isberg P.E., Gisslén M., Björkman P. Is low-level HIV-1 viraemia associated with elevated levels of markers of immune activation, coagulation and cardiovascular disease? HIV Med. 2019; 20(9): 571–80. https://doi.org/10.1111/hiv.12756
- Utay N.S., Hunt P.W. Role of immune activation in progression to AIDS. Curr. Opin. HIV AIDS. 2016; 11(2): 131–7. https://doi.org/10.1097/coh.0000000000000242
- Younas M., Psomas C., Reynes C., Cezar R., Kundura L., Portalès P., et al. Residual viremia is linked to a specific immune activation profile in HIV-1-infected adults under efficient antiretroviral therapy. Front. Immunol. 2021; 12: 663843. https://doi.org/10.3389/fimmu.2021.663843
- Fombellida-Lopez C., Berkhout B., Darcis G., Pasternak A.O. Persistent HIV-1 transcription during ART: time to reassess its significance? Curr. Opin. HIV AIDS. 2024; 19(3): 124–32. https://doi.org/10.1097/coh.0000000000000849
- Kuniholm J., Armstrong E., Bernabe B., Coote C., Berenson A., Patalano S.D., et al. Intragenic proviral elements support transcription of defective HIV-1 proviruses. PLoS Pathog. 2021; 17(12): e1009982. https://doi.org/10.1371/journal.ppat.1009982
- Marchetti G., Tincati C., Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 2013; 26(1): 2–18. https://doi.org/10.1128/cmr.00050-12
- Freeman M.L., Lederman M.M., Gianella S. Partners in Crime: The Role of CMV in immune dysregulation and clinical outcome during HIV infection. Curr. HIV/AIDS Rep. 2016; 13(1): 10–9. https://doi.org/10.1007/s11904-016-0297-9
- Sherman B.T., Hu X., Singh K., Haine L., Rupert A.W., Neaton J.D., et al. Genome-wide association study of high-sensitivity C-reactive protein, D-dimer, and interleukin-6 levels in multiethnic HIV+ cohorts. AIDS. 2021; 35(2): 193–204. https://doi.org/10.1097/qad.0000000000002738
- Shirley D.K., Kaner R.J., Glesby M.J. Effects of smoking on non-AIDS-related morbidity in HIV-infected patients. Clin. Infect. Dis. 2013; 57(2): 275–82. https://doi.org/10.1093/cid/cit207
- Rapid Response Service. Low-level HIV viremia: Definitions, predictors, mechanisms, and clinical outcomes. Toronto, ON: The Ontario HIV Treatment Network; 2022. Available at: https://www.ohtn.on.ca/wp-content/uploads/2022/01/RR166_Low-level-viremia_version2.pdf
- Wu F., Simonetti F.R. Learning from persistent viremia: mechanisms and implications for clinical care and HIV-1 cure. Curr. HIV/AIDS Rep. 2023; 20(6): 428–39. https://doi.org/10.1007/s11904-023-00674-w
- Bobkova M.R. Low-level viremia in HIV infection: causes and consequences. VICh-infektsiya i immunosupressii. 2024; 16(2): 7–22. https://doi.org/10.22328/2077-9828-2024-16-2-7-22 https://elibrary.ru/zlmcgr (in Russian)
- Genoyer E., Lopez C.B. The impact of defective viruses on infection and immunity. Annu. Rev. Virol. 2019; 6(1): 547–66. https://doi.org/10.1146/annurev-virology-092818-015652
- Wang H., Cui X., Cai X., An T. Recombination in positive-strand RNA viruses. Front. Microbiol. 2022; 13: 870759. https://doi.org/10.3389/fmicb.2022.870759
- Sanchez G., Xu X., Chermann J.C., Hirsch I. Accumulation of defective viral genomes in peripheral blood mononuclear cells of human immunodeficiency virus type 1-infected individuals. J. Virol. 1997; 71(3): 2233–40. https://doi.org/10.1128/jvi.71.3.2233-2240.1997
- Imamichi H., Dewar R.L., Adelsberger J.W., Rehm C.A., O’Doherty U., Paxinos E.E., et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl Acad. Sci. USA. 2016; 113(31): 8783–8. https://doi.org/10.1073/pnas.1609057113
- Kilroy J.M., Leal A.A., Henderson A.J. Chronic HIV transcription, translation, and persistent inflammation. Viruses. 2024; 16(5): 751. https://doi.org/10.3390/v16050751
- Berkhout B., van Wamel J., Klaver B. Requirements for DNA strand transfer during reverse transcription in mutant HIV-1 virions. J. Mol. Biol. 1995; 252(1): 59–69. https://doi.org/10.1006/jmbi.1994.0475
- Ho Y.C., Shan L., Hosmane N.N., Wang J., Laskey S.B., Rosenbloom D.I., et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013; 155(3): 540–51. https://doi.org/10.1016/j.cell.2013.09.020
- López CB. Defective viral genomes: critical danger signals of viral infections. J. Virol. 2014; 88(16): 8720–3. https://doi.org/10.1128/jvi.00707-14
- Vignuzzi M., Lopez C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019; 4(7): 1075–87. https://doi.org/10.1038/s41564-019-0465-y
- Kuniholm J., Coote C., Henderson A.J. Defective HIV-1 genomes and their potential impact on HIV pathogenesis. Retrovirology. 2022; 19(1): 13. https://doi.org/10.1186/s12977-022-00601-8
- Reeves D.B., Gaebler C., Oliveira T.Y., Peluso M.J., Schiffer J.T., Cohn L.B., et al. Impact of misclassified defective proviruses on HIV reservoir measurements. Nat. Commun. 2023; 14(1): 4186. https://doi.org/10.1038/s41467-023-39837-z
- Bobkova M.R. Cellular proteins as potential targets for antiretroviral therapy. Voprosy virusologii. 2023; 68(6): 488–504. https://doi.org/10.36233/0507-4088-207 https://elibrary.ru/klgwak (in Russian)
- Hadpech S., Moonmuang S., Chupradit K., Yasamut U., Tayapiwatana C. Updating on roles of HIV intrinsic factors: a review of their antiviral mechanisms and emerging functions. Intervirology. 2022; 65(2): 67–79. https://doi.org/10.1159/000519241
- Ramdas P., Sahu A.K., Mishra T., Bhardwaj V., Chande A. From entry to egress: strategic exploitation of the cellular processes by HIV-1. Front. Microbiol. 2020; 11: 559792. https://doi.org/10.3389/fmicb.2020.559792
- Nchioua R., Bosso M., Kmiec D., Kirchhoff F. Cellular factors targeting HIV-1 transcription and viral RNA transcripts. Viruses. 2020; 12(5): 495. https://doi.org/10.3390/v12050495
- Ghimire D., Rai M., Gaur R. Novel host restriction factors implicated in HIV-1 replication. J. Gen. Virol. 2018; 99(4): 435–46. https://doi.org/10.1099/jgv.0.001026
- Colomer-Lluch M., Ruiz A., Moris A., Prado J.G. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front. Immunol. 2018; 9: 2876. https://doi.org/10.3389/fimmu.2018.02876
- Schaller T., Herold N. The early bird catches the worm – can evolution teach us lessons in fighting HIV? Curr. HIV Res. 2016; 14(3): 183–210. https://doi.org/10.2174/1570162x14999160224094914
- Bedwell G.J., Engelman A.N. Factors that mold the nuclear landscape of HIV-1 integration. Nucleic Acids Res. 2021; 49(2): 621–35. https://doi.org/10.1093/nar/gkaa1207
- Craigie R., Bushman F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012; 2(7): a006890. https://doi.org/10.1101/cshperspect.a006890
- Rodgers K., McVey M. Error-prone repair of DNA double-strand breaks. J. Cell. Physiol. 2016; 231(1): 15–24. https://doi.org/10.1002/jcp.25053
- Wiegand A., Spindler J., Hong F.F., Shao W., Cyktor J.C., Cillo A.R., et al. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc. Natl Acad. Sci. USA. 2017; 114(18): E3659–68. https://doi.org/10.1073/pnas.1617961114
- Dutilleul A., Rodari A., Van Lint C. Depicting HIV-1 transcriptional mechanisms: a summary of what we know. Viruses. 2020; 12(12): 1385. https://doi.org/10.3390/v12121385
- Ding P., Kharytonchyk S., Waller A., Mbaekwe U., Basappa S., Kuo N., et al. Identification of the initial nucleocapsid recognition element in the HIV-1 RNA packaging signal. Proc. Natl Acad. Sci. USA. 2020; 117(30): 17737–46. https://doi.org/10.1073/pnas.2008519117
- Ishizaka A., Sato H., Nakamura H., Koga M., Kikuchi T., Hosoya N., et al. Short intracellular HIV-1 transcripts as biomarkers of residual immune activation in patients on antiretroviral therapy. J. Virol. 2016; 90(12): 5665–76. https://doi.org/10.1128/jvi.03158-15
- Sertznig H., Hillebrand F., Erkelenz S., Schaal H., Widera M. Behind the scenes of HIV-1 replication: Alternative splicing as the dependency factor on the quiet. Virology. 2018; 516: 176–88. https://doi.org/10.1016/j.virol.2018.01.011
- Mancarella A., Procopio F.A., Achsel T., De Crignis E., Foley B.T., Corradin G., et al. Detection of antisense protein (ASP) RNA transcripts in individuals infected with human immunodeficiency virus type 1 (HIV-1). J. Gen. Virol. 2019; 100(5): 863–76. https://doi.org/10.1099/jgv.0.001244
- Imamichi H., Smith M., Adelsberger J.W., Izumi T., Scrimieri F., Sherman B.T., et al. Defective HIV-1 proviruses produce viral proteins. Proc. Natl Acad. Sci. USA. 2020; 117(7): 3704–10. https://doi.org/10.1073/pnas.1917876117
- Vanhee-Brossollet C., Thoreau H., Serpente N., D’Auriol L., Levy J.P., Vaquero C. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology. 1995; 206(1): 196–202. https://doi.org/10.1016/s0042-6822(95)80034-4
- Pollack R.A., Jones R.B., Pertea M., Bruner K.M., Martin A.R., Thomas A.S., et al. Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host Microbe. 2017; 21(4): 494–506.e4. https://doi.org/10.1016/j.chom.2017.03.008
- Decout A., Katz J.D., Venkatraman S., Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021; 21(9): 548–69. https://doi.org/10.1038/s41577-021-00524-z
- Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010; 11(11): 997–1004. https://doi.org/10.1038/ni.1932
- Wu B., Hur S. How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 2015; 12: 91–8. https://doi.org/10.1016/j.coviro.2015.04.004
- Ferdin J., Goričar K., Dolžan V., Plemenitaš A., Martin J.N., Peterlin B.M., et al. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS One. 2018; 13(1): e0191613. https://doi.org/10.1371/journal.pone.0191613
- Fenwick C., Joo V., Jacquier P., Noto A., Banga R., Perreau M., et al. T-cell exhaustion in HIV infection. Immunol. Rev. 2019; 292(1): 149–63. https://doi.org/10.1111/imr.12823
- Verdon D.J., Mulazzani M., Jenkins M.R. Cellular and molecular mechanisms of CD8(+) T cell differentiation, dysfunction and exhaustion. Int. J. Mol. Sci. 2020; 21(19): 7357. https://doi.org/10.3390/ijms21197357
- Lichterfeld M., Gao C., Yu X.G. An ordeal that does not heal: understanding barriers to a cure for HIV-1 infection. Trends Immunol. 2022; 43(8): 608–16. https://doi.org/10.1016/j.it.2022.06.002
- Roux H., Chomont N. Measuring human immunodeficiency virus reservoirs: do we need to choose between quantity and quality? J. Infect. Dis. 2024; 229(3): 635–43. https://doi.org/10.1093/infdis/jiad381
- Halvas E.K., Joseph K.W., Brandt L.D., Guo S., Sobolewski M.D., Jacobs J.L., et al. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J. Clin. Invest. 2020; 130(11): 5847–57. https://doi.org/10.1172/jci138099
- Virgilio M.C., Collins K.L. The impact of cellular proliferation on the HIV-1 reservoir. Viruses. 2020; 12(2): 127. https://doi.org/10.3390/v12020127
- White J.A., Wu F., Yasin S., Moskovljevic M., Varriale J., Dragoni F., et al. Clonally expanded HIV-1 proviruses with 5’-leader defects can give rise to nonsuppressible residual viremia. J. Clin. Invest. 2023; 133(6): e165245. https://doi.org/10.1172/jci165245
- Bui J.K., Sobolewski M.D., Keele B.F., Spindler J., Musick A., Wiegand A., et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017; 13(3): e1006283. https://doi.org/10.1371/journal.ppat.1006283
- Maldarelli F., Wu X., Su L., Simonetti F.R., Shao W., Hill S., et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014; 345(6193): 179–83. https://doi.org/10.1126/science.1254194
- Linden N., Jones R.B. Potential multi-modal effects of provirus integration on HIV-1 persistence: lessons from other viruses. Trends Immunol. 2022; 43(8): 617–29. https://doi.org/10.1016/j.it.2022.06.001
- Mohammadi A., Etemad B., Zhang X., Li Y., Bedwell G.J., Sharaf R., et al. Viral and host mediators of non-suppressible HIV-1 viremia. Nat. Med. 2023; 29(12): 3212–23. https://doi.org/10.1038/s41591-023-02611-1
- Crespo-Bermejo C., de Arellano E.R., Lara-Aguilar V., Valle-Millares D., Gomez-Lus M.L., Madrid R., et al. Persistent low-Level viremia in persons living with HIV undertreatment: An unresolved status. Virulence. 2021; 12(1): 2919–31. https://doi.org/10.1080/21505594.2021.2004743
- Sannier G., Dubé M., Dufour C., Richard C., Brassard N., Delgado G.G., et al. Combined single-cell transcriptional, translational, and genomic profiling reveals HIV-1 reservoir diversity. Cell Rep. 2021; 36(9): 109643. https://doi.org/10.1016/j.celrep.2021.109643
- Brodin J., Zanini F., Thebo L., Lanz C., Bratt G., Neher R.A., et al. Establishment and stability of the latent HIV-1 DNA reservoir. Elife. 2016; 5: e18889. https://doi.org/10.7554/elife.18889
- Lian X., Gao C., Sun X., Jiang C., Einkauf K.B., Seiger K.W., et al. Signatures of immune selection in intact and defective proviruses distinguish HIV-1 elite controllers. Sci. Transl. Med. 2021; 13(624): eabl4097. https://doi.org/10.1126/scitranslmed.abl4097
- Abrahams M.R., Joseph S.B., Garrett N., Tyers L., Moeser M., Archin N., et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019; 11(513): eaaw5589. https://doi.org/10.1126/scitranslmed.aaw5589
- Lai M., Maori E., Quaranta P., Matteoli G., Maggi F., Sgarbanti M., et al. CRISPR/Cas9 ablation of integrated HIV-1 accumulates proviral DNA circles with reformed long terminal repeats. J. Virol. 2021; 95(23): e0135821. https://doi.org/10.1128/jvi.01358-21
Supplementary files

