



Vol 69, No 4 (2024)

- Year: 2024
- Published: 26.09.2024
- Articles: 10
- URL: https://virusjour.crie.ru/jour/issue/view/135
Full Issue

ORIGINAL RESEARCHES
Molecular detection of high-risk papillomaviruses and vaccination status in normal cytology in Congo
Abstract
Objective: The aim of this study was to identify the molecular prevalence of high-risk HPV infection and the distribution of genotypes present in normal cytology, as well as to determine the vaccination status of our study population.
Methods: 110 cervical samples were taken from individuals, and 1 ml of each sample was added to the Xpert HPV cartridge in the sample compartment before it was placed in the Cepheid GeneXpert system. Detection was performed simultaneously via amplification of the E6 and E7 genes in five fluorescent channels (HPV16, HPV18/45, HPV31/33/35/52/58, HPV51/59, and HPV39/56/66/68a).
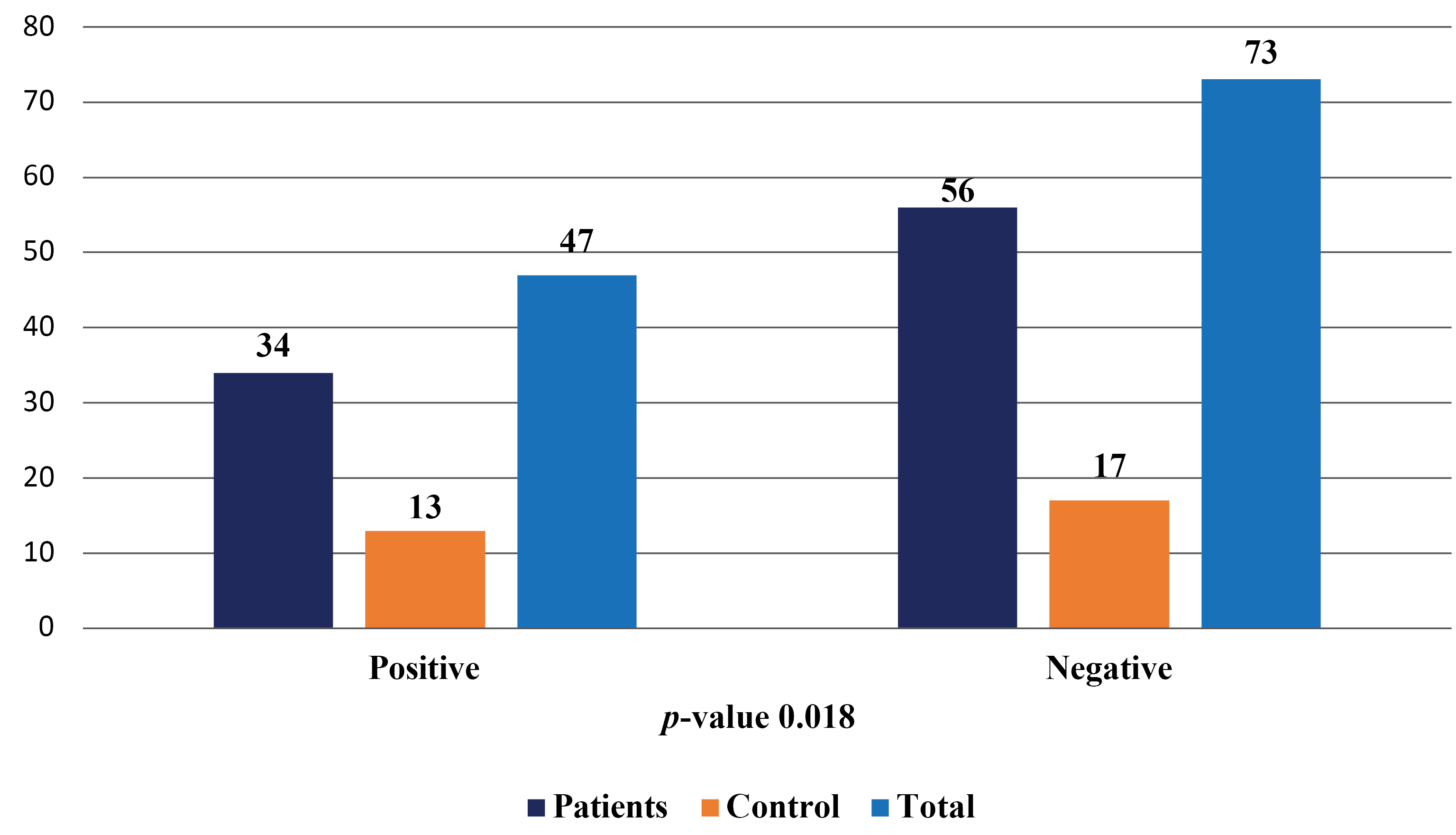
Results: 36/110 (33%) of all samples tested were positive for HPV DNA. The predominant genotypes were HPV16 (12.7%) and other pooled HR-HPV types (8.2%). All women who received the Gardasil-9 vaccine (3.6%) had HPV, and infection was associated with travel outside Africa. 96.4% of the screened individuals had not received any HPV vaccine.
Conclusion: Our research confirms a widespread HR-HPV infection in our population and extends the importance of studies on the molecular prevalence of HPV, particularly in women with normal cytology and apparent good health, in view of the cruel lack of public awareness of HPV infections.
 301-308
301-308



The effect of sodium deoxyribonucleate with iron complex on the expression of surface markers of MT-4 cells infected with human immunodeficiency virus type 1 (HIV-1) (Retroviridae: Primate lentivirus group)
Abstract
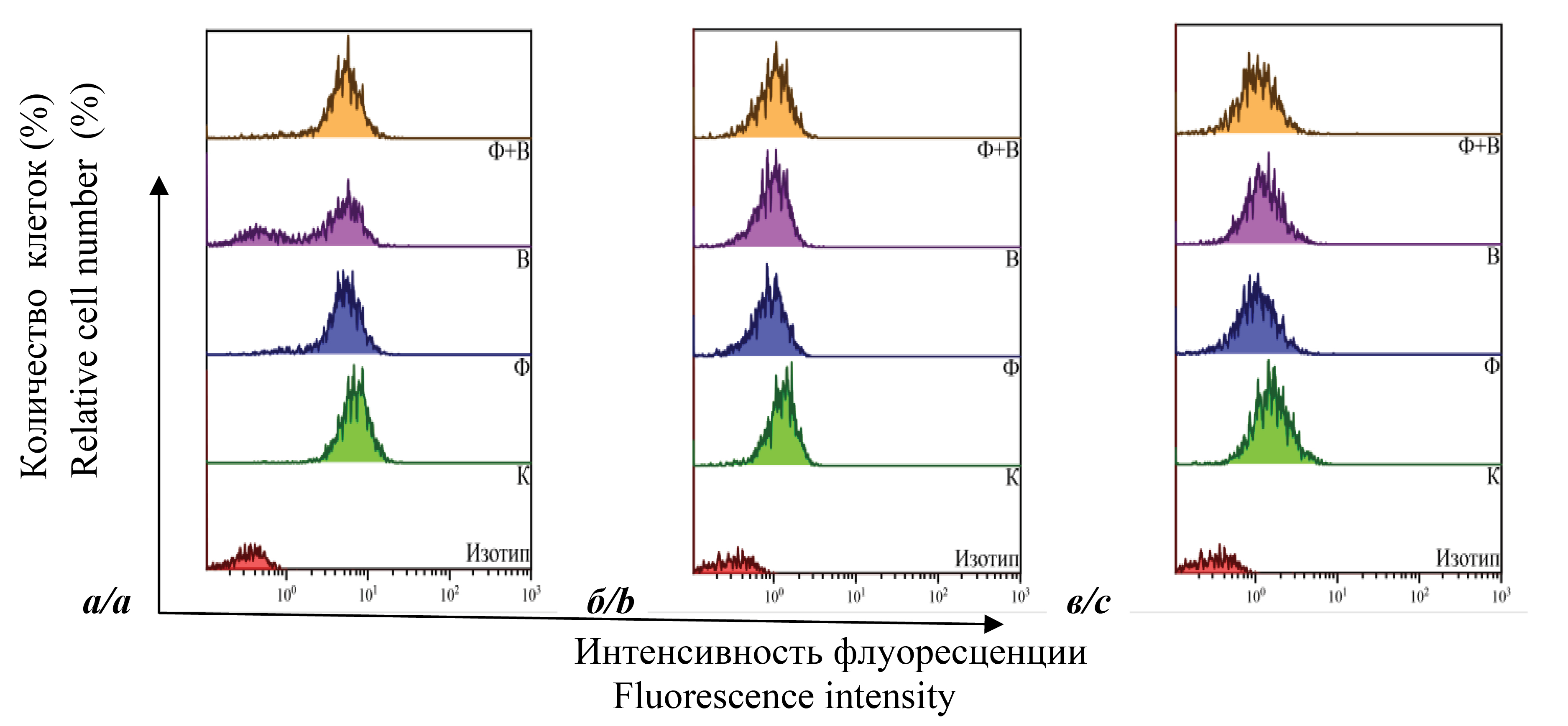
Introduction. The persistence of immune dysfunction during therapy has serious consequences for the health of HIV-infected people. Therefore, an important direction is the search for drugs that can reduce the inflammatory potential of the immune system and serve as an additional component of antiviral therapy.
Aim ‒ to study the effect of the immunomodulatory drug Sodium deoxyribonucleate with iron complex (DNA-Na-Fe) on the expression of activation markers in MT-4 cells infected with HIV-1.
Materials and methods. Expression levels of CD4, CD28, CD38, CD62L and HLA-DR proteins on the plasma membrane were measured in cells. To assess viral activity, the p24 protein was quantified by ELISA.
Results and discussion. The two cell variants with different replicative activity were analyzed. Control cells, cells with DNA-Na-Fe, infected cells and infected cells with DNA-Na-Fe were tested. Based on the results obtained, it can be concluded that antiviral activity of the drug in MT-4 cells infected with HIV-1 is associated with immunomodulatory activity that enhances the expression of membrane proteins CD4, CD28, CD38 and CD62L. Diversity in the effect of DNA-Na-Fe on the studied surface proteins expression in two cell lines indicates that they depend on the characteristics of the combined molecular biological processes occurring in cells. And the increased effects observed in a system with changes in replicative activity assumes its active participation in virus replication at the stages of virus penetration and budding.
Conclusion. Studies have shown that DNA-Na-Fe has antiviral and immunomodulatory activity.
309-319
Seropositivity of West Nile virus among acute febrile patients in Ilorin, Nigeria
Abstract
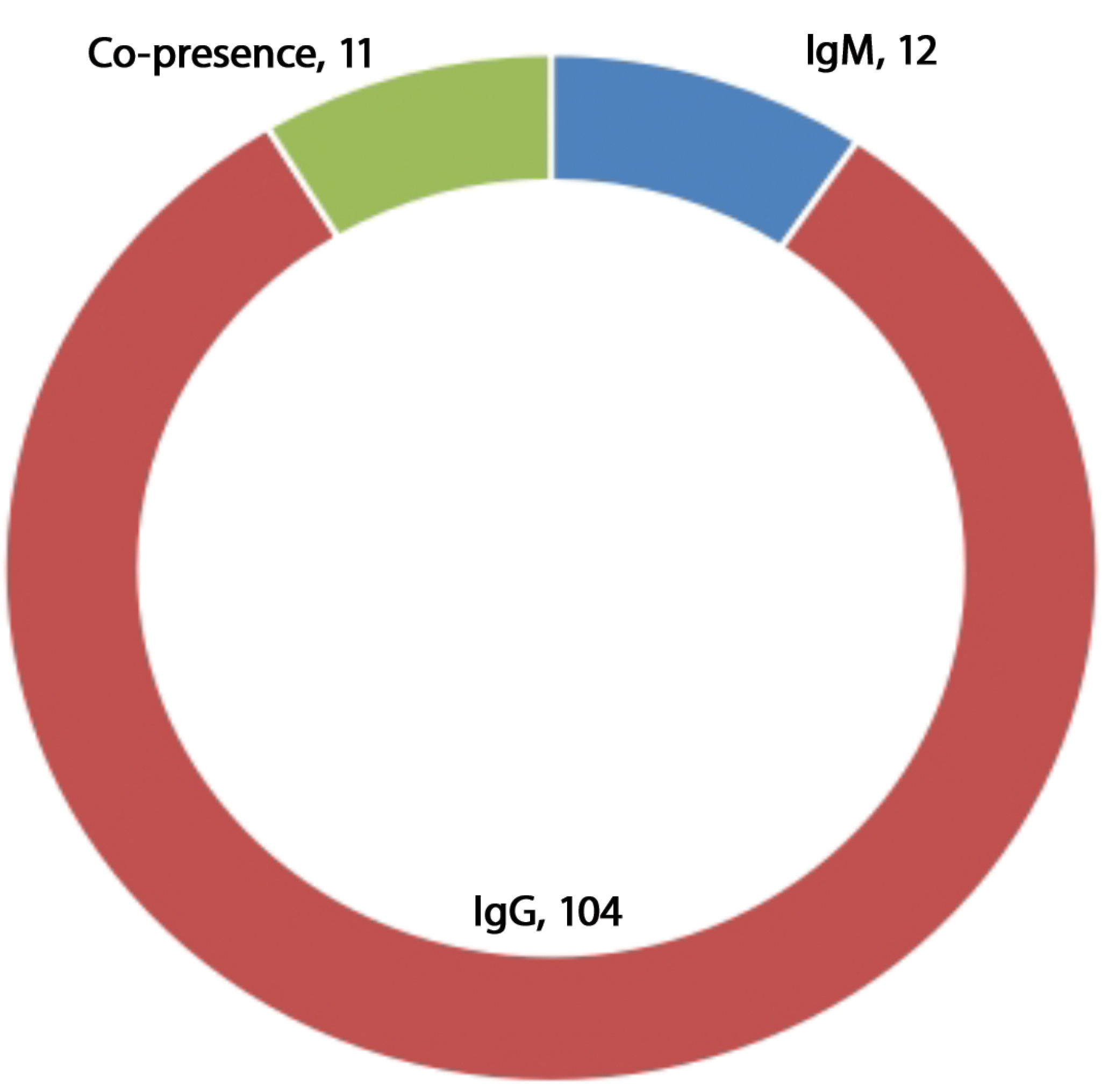
Introduction. West Nile Virus (WNV), a member of Flaviviridae family, is one of the most widely distributed arboviruses in the world. In developing countries like Nigeria, fever resulting from the WNV infection is often presumptively ascribed to malaria or typhoid due to misdiagnosis and low-level awareness of the viral infection. This study determined the prevalence of WNV IgM and IgG antibodies among febrile patients in the Ilorin metropolis.
Materials and methods. A total of two hundred (200) blood samples were collected from consenting patients and each serum was screened for anti-WNV IgM and IgG antibodies using indirect enzyme-linked immunosorbent assay (ELISA). Statistical correlation and logistic regression analysis were conducted.
Results. Overall, 6% (12/200) anti-WNV IgM seropositivity rate was recorded amongst the acute febrile patients with higher prevalence (6.30%) in females than in males (5.45%). Anti-WNV IgG positivity rate of 52% (104/200) was recorded, with 50.67% positivity rate in males and 38.95% in female participants. The convalescence phase posited by the 5.4% (11/200) co-detection of anti-WNV IgG and IgM antibodies among the participants was recorded. A statistical correlation was noticed with the age and religion of respondents to WNV serological positivity while gender, occupation, use of mosquito nets and formal education had no positive correlation at p < 0.05. However, based on odd ratio at 95% CI and logistic regression coefficients, the evaluated risk factors such as blood transfusion, residency, malaria parasite, and proximity to stagnant water and bush were significant to anti-WNV IgG and IgM positivity.
Conclusion. The findings of this study show the circulation of WNV in the study area. There is an urgent need for clinicians/physicians to include screening for the West Nile virus in cases of febrile patients before the commencement of treatment.
320-328
The Molecular and Biological Patterns Underlying Sustained SARS-CoV-2 Circulation in the Human Population
Abstract
Introduction. For four years, SARS-CoV-2, the etiological agent of COVID-19, has been circulating among humans. By the end of the second year, an absence of immunologically naive individuals was observed, attributable to extensive immunization efforts and natural viral exposure. This study focuses on delineating the molecular and biological patterns that facilitate the persistence of SARS-CoV-2, thereby informing predictions on the epidemiological trajectory of COVID-19 toward refining pandemic countermeasures.
The aim of this study was to describe the molecular biological patterns identified that contribute to the persistence of the virus in the human population.
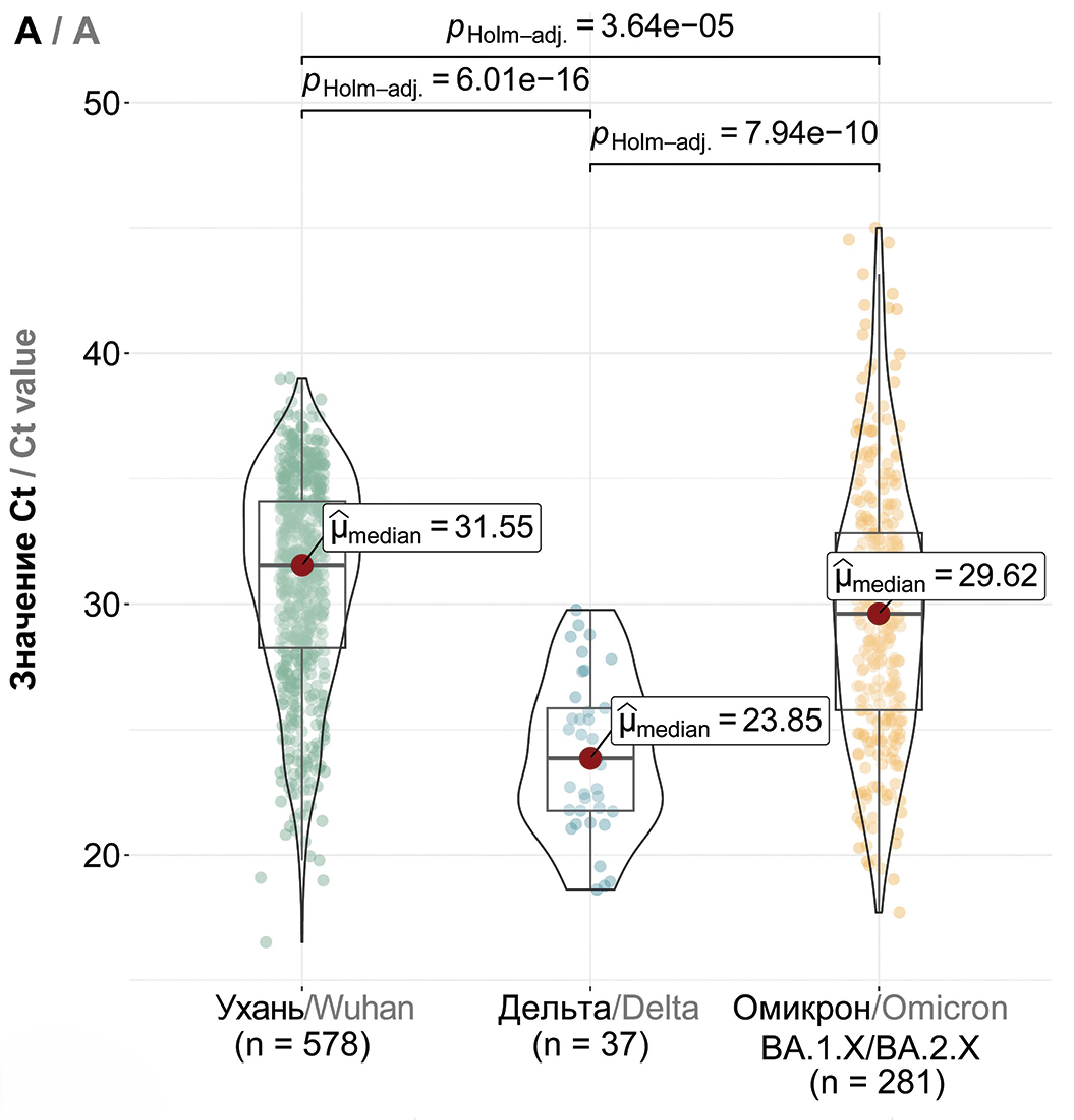
Materials and methods. For over three years since the beginning of the COVID-19 pandemic, molecular genetic monitoring of SARS-CoV-2 has been conducted, which included the collection of nasopharyngeal swabs from infected individuals, assessment of viral load, and subsequent whole-genome sequencing.
Results. We discerned dominant genetic lineages correlated with rising disease incidence. We scrutinized amino acid substitutions across SARS-CoV-2 proteins and quantified viral loads in swab samples from patients with emerging COVID-19 variants. Our findings suggest a model of viral persistence characterized by 1) periodic serotype shifts causing substantial diminutions in serum virus-neutralizing activity (> 10-fold), 2) serotype-specific accrual of point mutations in the receptor-binding domain (RBD) to modestly circumvent neutralizing antibodies and enhance receptor affinity, and 3) a gradually increasing amount of virus being shed in mucosal surfaces within a single serotype.
Conclusion. This model aptly accounts for the dynamics of COVID-19 incidence in Moscow. For a comprehensive understanding of these dynamics, acquiring population-level data on immune tension and antibody neutralization relative to genetic lineage compositions is essential.
329-340
Development, study, and comparison of models of cross-immunity to the influenza virus using statistical methods and machine learning
Abstract
Introduction. The World Health Organization considers the values of antibody titers in the hemagglutination inhibition assay as one of the most important criteria for assessing successful vaccination. Mathematical modeling of cross-immunity allows for identification on a real-time basis of new antigenic variants, which is of paramount importance for human health.
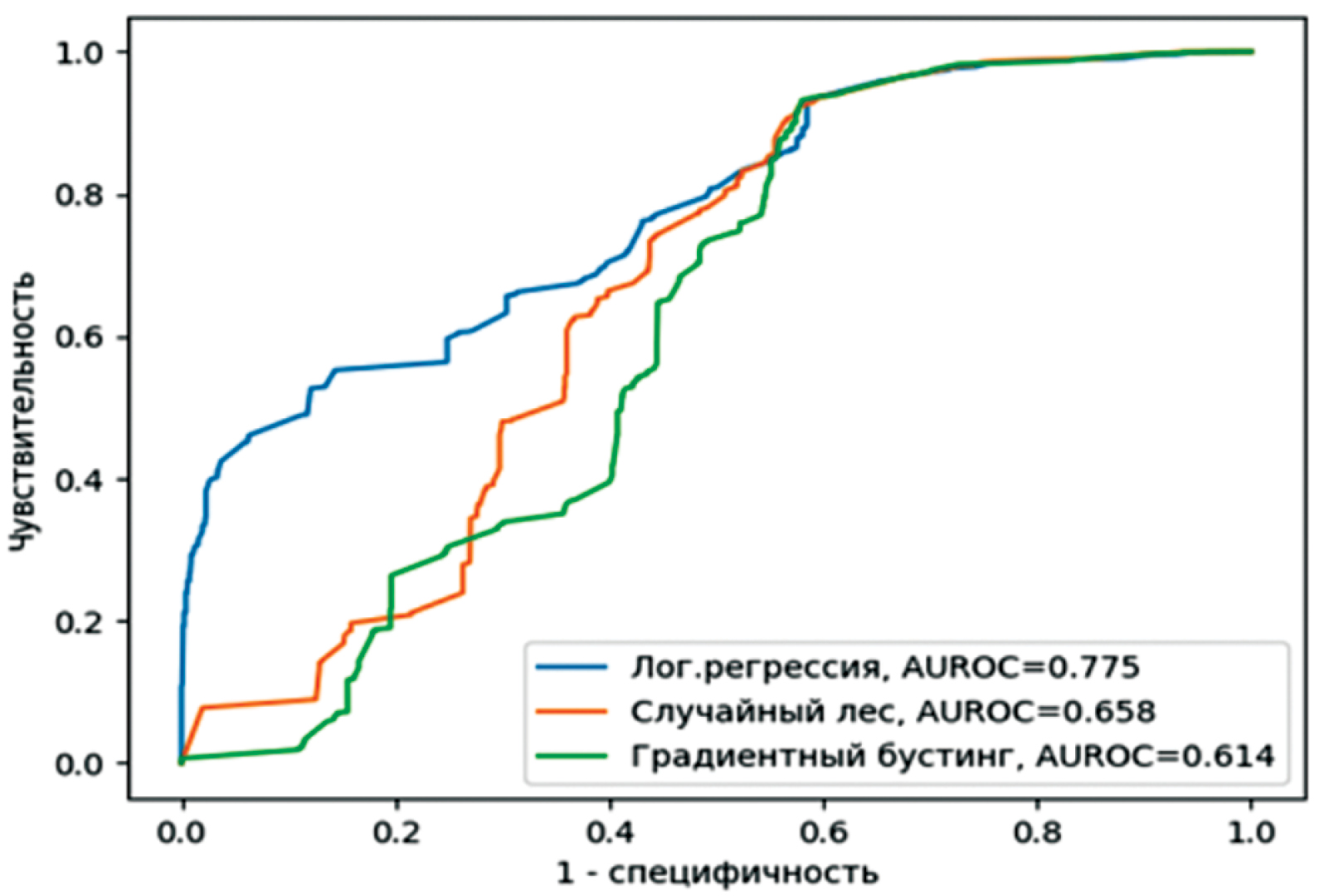
Materials and methods. This study uses statistical methods and machine learning techniques from simple to complex: logistic regression model, random forest method, and gradient boosting. The calculations used the AAindex matrices in parallel to the Hamming distance. The calculations were carried out with different types and values of antigenic escape thresholds, on four data sets. The results were compared using common binary classification metrics.
Results. Significant differentiation is shown depending on the data sets used. The best results were demonstrated by all three models for the forecast autumn season of 2022, which were preliminary trained on the February season of the same year (Auroc 0.934; 0.958; 0.956, respectively). The lowest results were obtained for the entire forecast year 2023, they were set up on data from two seasons of 2022 (Aucroc 0.614; 0.658; 0.775). The dependence of the results on the types of thresholds used and their values turned out to be insignificant. The additional use of AAindex matrices did not significantly improve the results of the models without introducing significant deterioration.
Conclusion. More complex models show better results. When developing cross-immunity models, testing on a variety of data sets is important to make strong claims about their prognostic robustness.
349-362
Molecular Study of Varicella zoster virus in Cerebrospinal Fluid from Stroke Patients of Thi-Qar province
Abstract
Introduction: Varicella zoster virus (VZV) is a type of alpha-herpesvirus that specifically targets the nervous system. The initial infection, typically occurring during childhood, results in varicella (commonly known as chickenpox), after which the virus enters a dormant state in cranial nerve ganglia, dorsal root ganglia, and autonomic ganglia throughout the entire neuroaxis.
Aim of the study: Molecular and genetic studies of viruses are an important tool for virus development and identifying viral treatments to combat the diseases. The aim of the study was to determine the whole ORF4 sequence of the local VZV strains for phylogenetic analysis to determine the variability in the viral sequence.
Material and methods: Ten samples of VZV DNA were subjected to the sequencing of the whole ORF4 region following identification using the PCR method.
Results: Sequences from five samples have been successfully analyzed. All clinical strains were discovered to possess a genome with a length of 124,884 base pairs. The sequences exhibited the occurrence of two distinct mutations, one being a transversion and the other a transition, with the latter resulting in an alteration of the amino acid. A phylogenetic tree was constructed using the maximum likelihood method based on the sequences of five nucleotide sequences from clinical samples and nine reference VZV strains. The tree displayed the evolutionary distances between these sequences. The analysis of the phylogenetic tree revealed the presence of five primary clades, with four of them originating from India (isolates S1, S2, S4, S5), while S3 exhibited similarity to a strain from the United Kingdom.
341-348
Unusual BA222-like strains of Rotavirus A (Sedoreoviridae: Rotavirus: Rotavirus A): molecular and genetic analysis based on all genome segments
Abstract
Introduction. Rotavirus infection is the major cause of severe dehydrating diarrhea requiring hospitalization in young children worldwide. Due to their segmented genome, rotaviruses are capable of gene reassortment, which makes the emergence and spread of genetically novel strains possible. The purpose of this study was to search for unusual rotaviruses circulating in Nizhny Novgorod in 2021‒2023 and their molecular genetic characterization based on all genome segments.
Materials and methods. Rotavirus-positive stool samples of children were examined by PCR genotyping and electrophoresis in PAAG. cDNA fragments of each of the 11 genes (VP1‒VP4, VP6, VP7, NSP1‒NSP5), 570 to 850 nucleotide pairs in length were sequenced for the selected strains. The phylogenetic analysis was performed in the MEGA X program.
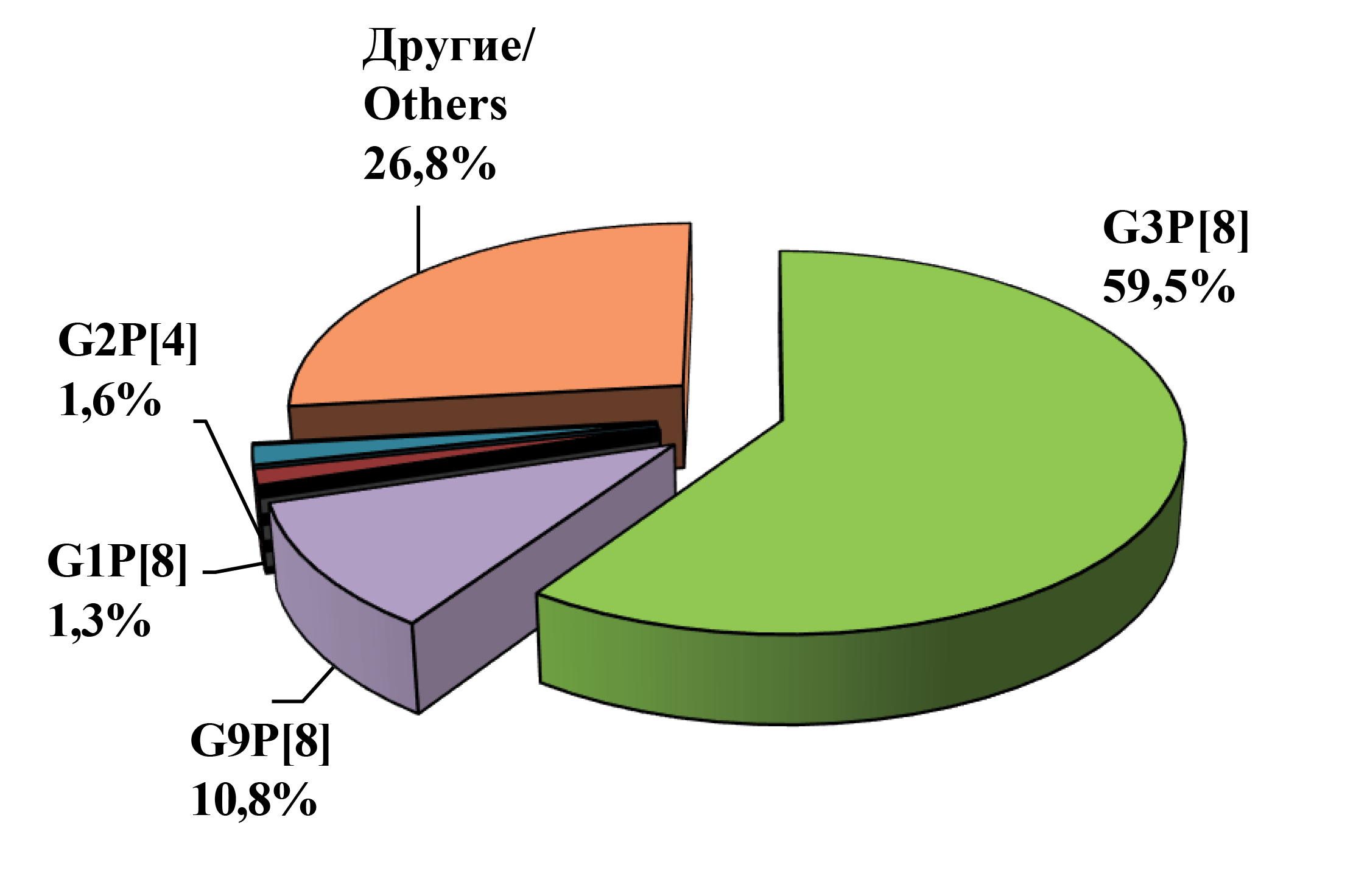
Results. In the study period 2021‒2023, 11 G[P] combinations with a predominance of G3P[8] (59.5%) were identified. Six atypical Rotavirus А (RVA) strains were identified: 2 strains of the G2P[4] genotype (G2-P[4]-I2-R2-C2-M2-A3-N2-T3-E2-H3, G2-P[4]-I2-R2-C2-M2-A3-N2-T3-E3-H2) and 4 G3P[9] strains (all strains had the genotype G3-P[9]-I2-R2-C2-M2-A3-N2-T3-E3-H3). Phylogenetic analysis based on all genes showed an evolutionary relationship between rotaviruses similar to rotaviruses of cats and dogs (BA222-like) and unusual strains of the G2P[4] genotype, for which a mixed combination of genotypes was identified and characterized for the first time.
Discussion. The results obtained expand the understanding of the diversity of reassortant RVAs, as well as complement the data on the genotypic structure of the rotavirus population in Nizhny Novgorod.
Conclusion. The wide genetic diversity of reassortant RVA can help rotaviruses overcome the immunological pressure provided by natural and vaccine-induced immunity. In this regard, to control the emergence of new variants and assess changes in the virulence of rotaviruses after reassortment processes, continuous molecular monitoring for circulating RVA is necessary.
363-376
First detection of influenza A virus subtypes H1N1 and H3N8 in the Antarctic region: King George Island, 2023
Abstract
Relevance. Influenza A virus is characterized by a segmented single-stranded RNA genome. Such organization of the virus genome determines the possibility of reassortment, which can lead to the emergence of new virus variants. The main natural reservoir of most influenza A virus subtypes are wild waterfowl. Seasonal migrations gather waterfowl from all major migration routes to nesting areas near the northern and southern polar circles. This makes intercontinental spread of influenza A viruses possible.
Objective ‒ to conduct molecular genetic monitoring and study the phylogenetic relationships of influenza A virus variants circulating in Antarctica in 2023.
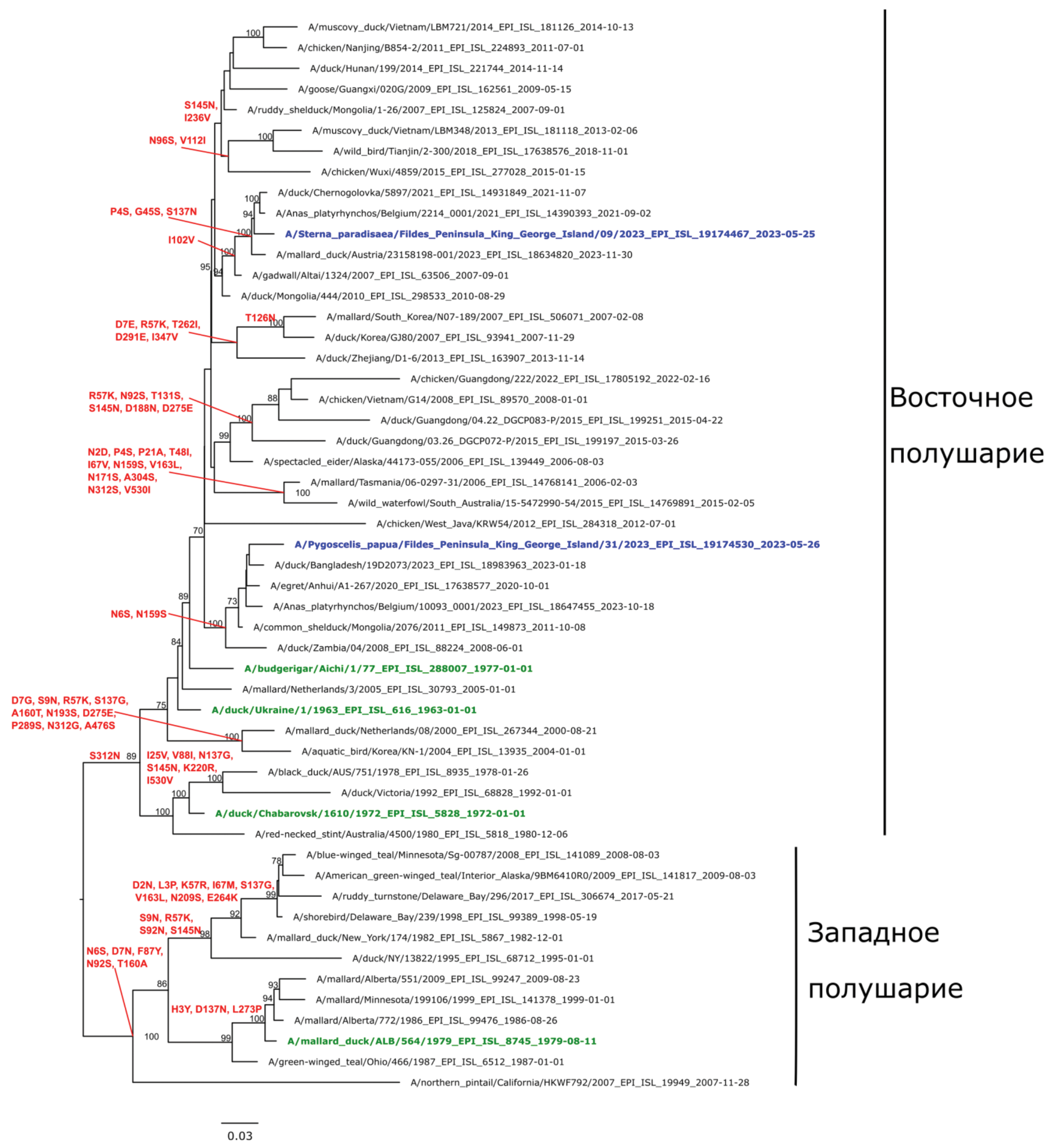
Materials and methods. We studied 84 samples of biological material obtained from birds and marine mammals in April‒May 2023 in coastal areas of Antarctica. For 3 samples, sequencing was performed on the Miseq, Illumina platform and phylogenetic analysis of the obtained nucleotide sequences of the influenza A virus genomes was performed.
Results. The circulation of avian influenza virus in the Antarctic region was confirmed. Heterogeneity of the pool of circulating variants of the influenza A virus (H3N8, H1N1) was revealed. Full-length genomes of the avian influenza virus were sequenced and posted in the GISAID database (EPI_ISL_19032103, 19174530, 19174467).
Conclusion. The study of the genetic diversity of influenza A viruses circulating in the polar regions of the Earth and the identification of the conditions for the emergence of new genetic variants is a relevant task for the development of measures to prevent biological threats.
377-389
BOOK REVIEW
Review of the collective monograph edited by Academician of the Russian Academy of Sciences V.I. Zlobin «Tick-borne encephalitis in the 21st century»
 390-391
390-391
OBITUARY
Lyudmila V. Kolobukhina
 392-392
392-392
Регистрационный номер и дата принятия решения о регистрации СМИ: серия ПИ № ФС77-77676 от 29.01.2020.