



First detection of influenza A virus subtypes H1N1 and H3N8 in the Antarctic region: King George Island, 2023
- Authors: Ohlopkova O.V.1,2, Goncharov A.E.3,4, Aslanov B.I.4, Fadeev A.V.5, Davidyuk Y.N.6, Moshkin A.D.2, Stolbunova K.A.2, Stepanyuk M.A.2, Sobolev I.A.2, Tyumentseva M.A.1, Tyumentsev A.I.1, Shestopalov A.M.2, Akimkin V.G.1
-
Affiliations:
- Central Research Institute of Epidemiology, Rospotrebnadzor
- Federal Research Center for Fundamental and Translational Medicine
- Institute of Experimental Medicine
- Northwestern State Medical University named after I.I. Mechnikov
- A.A. Smorodintsev Research Institute of Influenza, Ministry of Health of the Russian Federation
- Federal State Educational Institution of Higher Education «Kazan (Volga Region) Federal University»
- Issue: Vol 69, No 4 (2024)
- Pages: 377-389
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16674
- DOI: https://doi.org/10.36233/0507-4088-257
- EDN: https://elibrary.ru/qujzfv
- ID: 16674

Cite item

Abstract
Relevance. Influenza A virus is characterized by a segmented single-stranded RNA genome. Such organization of the virus genome determines the possibility of reassortment, which can lead to the emergence of new virus variants. The main natural reservoir of most influenza A virus subtypes are wild waterfowl. Seasonal migrations gather waterfowl from all major migration routes to nesting areas near the northern and southern polar circles. This makes intercontinental spread of influenza A viruses possible.
Objective ‒ to conduct molecular genetic monitoring and study the phylogenetic relationships of influenza A virus variants circulating in Antarctica in 2023.
Materials and methods. We studied 84 samples of biological material obtained from birds and marine mammals in April‒May 2023 in coastal areas of Antarctica. For 3 samples, sequencing was performed on the Miseq, Illumina platform and phylogenetic analysis of the obtained nucleotide sequences of the influenza A virus genomes was performed.
Results. The circulation of avian influenza virus in the Antarctic region was confirmed. Heterogeneity of the pool of circulating variants of the influenza A virus (H3N8, H1N1) was revealed. Full-length genomes of the avian influenza virus were sequenced and posted in the GISAID database (EPI_ISL_19032103, 19174530, 19174467).
Conclusion. The study of the genetic diversity of influenza A viruses circulating in the polar regions of the Earth and the identification of the conditions for the emergence of new genetic variants is a relevant task for the development of measures to prevent biological threats.
Full Text
Introduction
Influenza A virus belongs to the Orthomyxoviridae family and is characterized by a segmented single-stranded RNA genome. This organization of the virus genome determines the possibility of reassortment, which can lead to the emergence of new virus variants [1]. The main natural reservoir of orthomyxoviruses, including most subtypes of influenza viruses, is colonial seabirds [2‒5]. The ability of birds of this group to make long migrations, along with their mass density within colonies, contributes to the transcontinental spread of influenza A virus subtypes and expands the possibilities of transmission to different animal species, which, in turn, may contribute to the emergence of new variants and species of viruses due to recombination and reassortment of their genomes [1, 6‒8].
Due to a combination of special physiographic and ecological conditions, the circumpolar regions of the planet are points of attraction for millions of migratory seabirds and mammals, representing an active zone of interspecific contacts [9‒13]. The circulation of the influenza A virus in natural Antarctic and Arctic biocenoses has been known since the mid-1970s of the last century. The virus was isolated here from birds and cetaceans. Avian influenza viruses were first identified in the Antarctic region in 1976 ‒ 14 strains of influenza A virus subtype H1N3 were isolated from lung and liver tissues of whales of the Balaenopteridae family [14]. In the Arctic part of Canada in 1984‒1997, antibodies to influenza A viruses were detected in 1.2% of beluga whales using serologic methods [15]. Later in the early 2000s, avian influenza virus was detected in the Antarctic birds [16].
One of the polar territories with significant biodiversity is the South Shetland Islands located to the north of the Antarctic Peninsula. The largest of the islands in this archipelago is King George Island (Waterloo).
The island is home to a number of polar research stations belonging to Argentina (Carlini), Brazil (Comandante Ferras), China (Changcheng ‒ Great Wall), Poland (Henryk Arktovsky), Russia (Bellingshausen), Uruguay (General Artigas), Peru (Machu Picchu), Chile (President Eduardo Frei) and South Korea (King Sejong). The island also has tourism potential, with short excursions regularly organized for dozens of tourists arriving by sea or air. The island is home to 12 species of birds, including Lonnberg’s skuas (Catharacta antarctica lonnbergi), south polar skuas (Catharacta maccormicki), white plover (Chionis alba), cape petrel (Daption capense), Dominican Gull (Larus dominicanus), Southern Giant Petrel (Macronectes giganteus), Wilson’s Teal (Oceanites oceanicus), Black-bellied Teal (Fregetta tropica), Adelie Penguin (Pygoscelis adeliae), Antarctic Penguin (P. antarctica), Papua penguin (P. papua) and Antarctic tern (Sterna vittata) [17, 18]. Of mammal species, Weddell seals (Leptonychotes weddellii) and southern sea elephants (Mirounga leonina) are the most abundant. The area is also visited by Antarctic fur seals (Arctocephalus gazella) and sea leopards (Hydrurga leptonyx) [19].
Thus, this area has a combination of unique factors including abundance of nesting sites of long-distance migratory birds, large populations of potentially susceptible wild animals ‒ endemics, rotating groups of tourists and personnel of polar stations.
In 2011, the circulation of influenza A virus on the Antarctic continent was detected for the first time. The virus had the H4N7 subtype and was obtained from biomaterial from a giant petrel. Also this year, two samples of the H6N8 subtype were identified from an Antarctic skua and an Antarctic penguin [16]. In 2013, the Respiratory Virus Laboratory of the Oswaldo Cruz Institute (Brazil) collected 95 avian fecal samples from penguin colonies in the South Shetland Islands, near Antarctica, and tested them by reverse transcription polymerase chain reaction (RT-PCR) for detection of avian influenza virus. As a result, 4 full-length genomes of influenza A virus subtype H11N2 were obtained. The biomaterial was the fecal matter of Adelie penguins [16]. In 2015, the World Health Organization (WHO) Collaborating Center for Influenza Research in Melbourne, Australia, received samples from Antarctic penguins in which it was possible to detect the H5N5 subtype. In 2017, several samples of the H11N2 subtype were also obtained [20]. At the same time, the GISAID database does not contain information about influenza A virus subtypes circulating in the Antarctic region between 2018 and 2022. The first media reports of the highly pathogenic avian influenza of the H5N1 subtype reaching the Antarctic region came in October 20231. The virus was detected on the sub-Antarctic islands, specifically in South Georgia and the South Sandwich Islands, about 1,600 kilometers from the Antarctic mainland. It has also reached the Falkland Islands. Gulls, skuas and terns were the first to die en masse from avian influenza, then the disease spread to albatrosses, penguins and the Antarctic grouse. Influenza A subtype H5N1 was also detected in mammals – sea elephants and harbor seals were affected. A total of 18 isolates of subtype H5N1 from a total of 21 sequenced avian influenza samples from Antarctica are represented in the GISAID database for 2023. Influenza A subtypes identified in 2023 besides H5N1 are H3N8 (2 samples) and H1N1 (1 sample) [21].
Thus, due to the high variability of influenza viruses, WHO constantly emphasizes the necessity for global surveillance to detect genetic alterations in the influenza A virus genome, especially in relation to widely circulating subtypes, such as H3N8. Therefore, monitoring for avian influenza pathogens in the Antarctic territories is an urgent necessity.
The aim of the study was to identify genetic variants of influenza A virus circulating in the coastal territories of Antarctica and to determine the characteristics of their genome.
Materials and methods
The biological material for the study (feces, excretions of birds and marine mammals, 84 samples in total) was collected during the route works conducted within the Fildes Peninsula of King George Island in April‒May 2023. Species composition of the surveyed representatives of the Antarctic fauna was as follows: Black-bellied Buzzard (9 positive samples in influenza A RNA detection), Antarctic Petrel (1 positive sample), Thick-billed Prion (3 positive samples), Arctic Tern (2 positive samples), Antarctic Cormorant (1 positive sample), Snow Petrel (7 positive samples), Papua penguin (3 positive specimens), Dominican gull (2 positive specimens), sea elephant (6 positive specimens), white plover (1 positive specimen), southern giant petrel (1 positive specimen), south polar skua (7 positive specimens), Adelie penguin (3 positive specimens).
Total RNA extraction was performed using a kit for RNA isolation from animal/bacterial cells, epithelial cell smear/scrape, and viruses on columns from Biolabmix LLC (Russia). Avian influenza RNA detection was carried out by RT-PCR with detection of results by gel electrophoresis using the BioMaster RT-PCR-Premium (2×) kit (Biolabmix LLC, Russia).
Sequencing of the full-length genome of avian influenza virus was performed using reagent kits from BioLink LLC (Russia): kit for enzymatic fragmentation of nucleic acids; reagent kit for preparation of NGS libraries for Illumina platform. The complementary DNA was purified on VAHTS DNA Clean Beads magnetic particles for DNA purification, (Vazyme, China). Genomic libraries were sequenced on the MiSeq platform, Illumina using MiSeq Reagent Kit v3 600-cycle. The BWA algorithm was used to align the obtained reads to a reference sequence. Samtools and Ivar tools were used to obtain consensus sequences.
A preliminary phylogenetic analysis was performed to compile a sample of sequences. A total of 657 isolates of H1N1 subtype and 1908 isolates of H3N8 subtype from the GISAID EpiFLU database were used to construct primary phylogenetic trees using the IQTree2 algorithm in the automatic parameter selection mode. Samples were further reduced to 50 sequences using the PARNAS tool. Comparative phylogenetic analysis of nucleotide sequences was performed by the maximum likelihood method using the RAxML algorithm [22] and the GTRGAMMA nucleotide substitution model. Statistical support for the branches of the tree was determined by the rapid bootstrap method with 1000 replications. The phylogenetic tree was midpoint rooting. Ancestral sequences were reconstructed using the RAxML algorithm [23]. Phylogenetic trees were visualized and annotated using Figtree and Inkscape tools. Amino acid substitutions in sequences were analyzed using MEGA and SnapGene software.
Results
Eighty-four samples of biological material were analyzed by RT-PCR for detection of influenza A virus RNA. Of these, 46 promising samples were selected for full-genome sequencing. According to the results of comparative analysis of the obtained nucleotide sequences and phylogenetic analysis, the studied samples were identified as influenza A virus subtypes H1N1 (1 sample) and H3N8 (2 samples).
The genome of the influenza A virus is known to encode 12 proteins. Of these, the genes of hemagglutinin and neuraminidase surface antigens, as well as the nonstructural protein NS1, responsible for interferon inhibition and characterized by considerable diversity among avian influenza strains, are of greatest interest for comparative and phylogenetic analysis [24].
H1N1 is a subtype of influenza A virus that is one of the most prevalent viruses in the world and was responsible for the 1918, 1977 and 2009 pandemics [25].
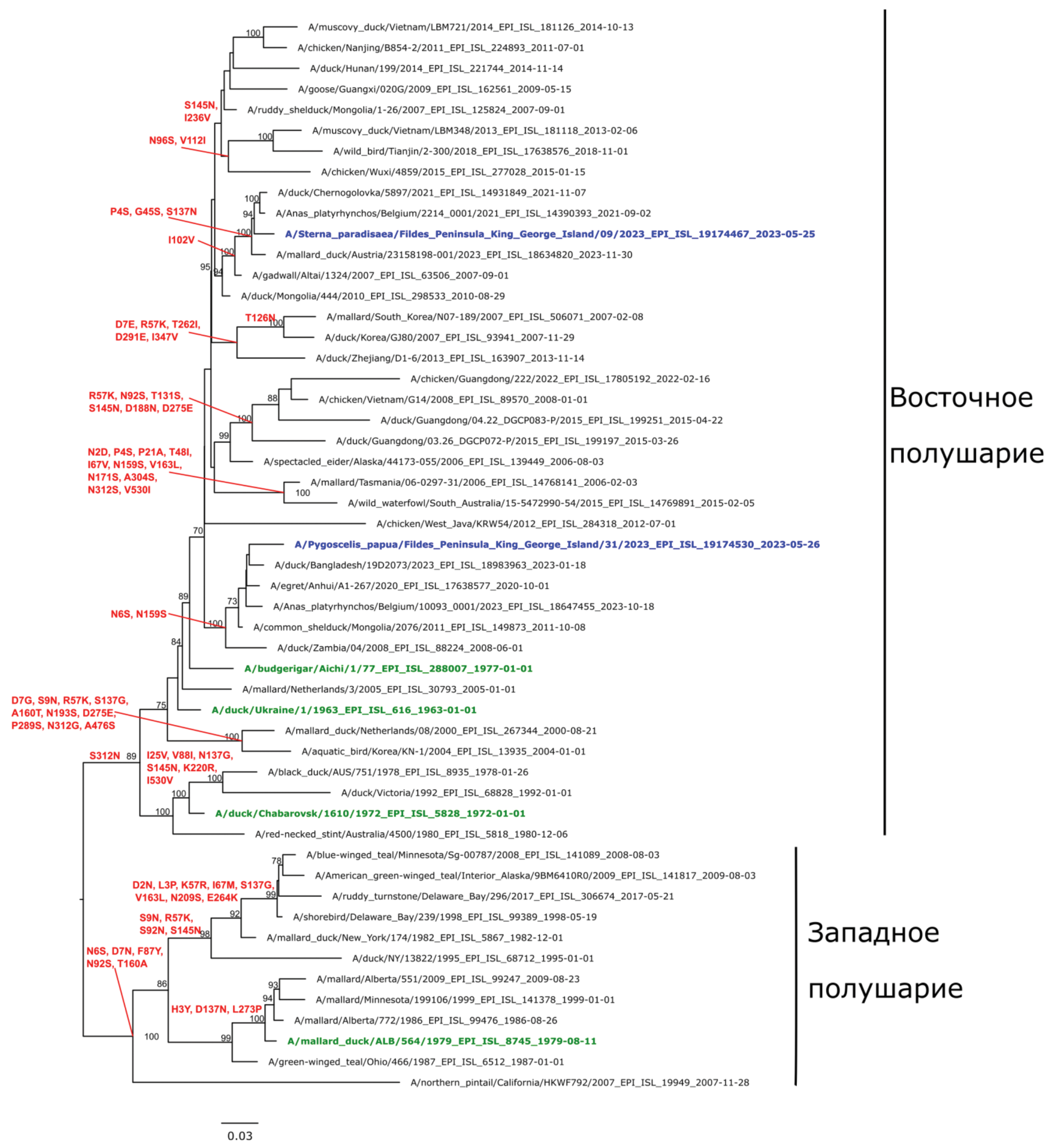
The phylogenetic environment of the H1N1 genome for the hemagglutinin gene identified by sequencing (Fig. 1) consists of two geographic groups: Asian and European, with two subgroups in the European group: European Russia (Moscow Region), European countries (Belgium, Netherlands) and the Asian part of Russia (Novosibirsk Region), Asian countries (Korea, China, Bangladesh, Mongolia). Phylogenetic analysis of the hemagglutinin gene of the studied H1N1 sequence revealed a close relationship with hemagglutinin genes of viruses found in ducks in Belgium and the Asian part of Russia, the similarity level was 97‒98%. At the same time, the most closely related were the strains that circulated mainly in wild waterfowl in the period 2018‒2023 (Fig. 1). In general, the nucleotide sequence of the hemagglutinin gene of the studied sample is characteristic of avian isolates of the Eastern Hemisphere.

Fig. 1. Phylogenetic tree for the hemagglutinin gene for influenza A virus subtype H1N1 isolates. Here and in Fig. 2‒5: The phylogenetic tree was constructed by the RAxML algorithm using the maximum likelihood (ML) method and the GTRGAMMA nucleotide substitution model. Statistical support for tree branches was determined by the rapid bootstrap method with 1000 replications. Support values below 70 are not shown. The phylogenetic tree is midpoint rooted. Ancestral sequences were reconstructed using the RAxML algorithm. Blue color on the trees indicates the sequences from this study. The oldest strains for genetic groups by hemagglutinin are shown in green. Substitutions in the corresponding reconstructed amino acid sequences are shown in red above the tree nodes.
Рис. 1. Филогенетическое дерево по гену гемагглютинина для изолятов вируса гриппа A субтипа H1N1. Здесь и на рис. 2‒5: филогенетическое дерево построено с помощью алгоритма RAxML методом максимального правдоподобия (ML) с использованием модели нуклеотидных замен GTRGAMMA. Статистическую поддержку ветвей дерева определяли методом быстрого бутстрепа (rapid bootstrap) c 1000 репликаций. Значения поддержки ниже 70 не показаны. Филогенетическое дерево укоренено на среднюю точку (midpoint rooting). Реконструкцию предковых последовательностей проводили с использованием алгоритма RAxML. Синим цветом на деревьях указаны последовательности образцов, секвенированные в ходе данного исследования. Зеленым цветом выделены старейшие штаммы для генетических групп по гемагглютинину. Красным цветом над узлами дерева обозначены замены в соответствующих реконструированных аминокислотных последовательностях.
On the phylogenetic tree of the neuraminidase gene (Fig. 2), the identified H1N1 strain has a position on the tree close to the isolates circulating in the territories of such countries as Belgium, the Netherlands, the European part of Russia (Moscow Region), in the period 2016‒2023, as well as in the territories of Belgium, South Korea and Novosibirsk region from 2019 to 2023. Moreover, the closest (Fig. 2) were A/Anas_platyrhynchos/Belgium/00358_0006/2023 and A/mallard/Novosibirsk_region/3445k/2020 with a difference of 3‒4% relative to the studied H1N1 isolate.

Fig. 2. Phylogenetic tree for the neuraminidase gene for influenza A virus subtype H1N1 isolates.
Рис. 2. Филогенетическое дерево по гену нейраминидазы для изолятов вируса гриппа A субтипа H1N1.
Influenza A virus of subtype H3N8 belongs to the subtypes most frequently encountered in birds. Interspecies transmission of the H3N8 subtype has been reported in various mammalian species, with endemic transmission in dogs and horses. To date, 3 cases of human infection with avian influenza A virus subtype H3N8 have been reported; all cases were reported in China between 2022 and 2023, with infection presumably resulting from direct or indirect contact with infected poultry [26].
The phylogenetically identified H3N8 subtype isolates (EPI_ISL_19174530 and EPI_ISL_ 19174467) have a distinct position relative to each other in terms of the gene encoding hemagglutinin. This is probably due to their independent evolution in the area of distribution. In general, the phylogenetic environment for the H3N8 isolate (EPI_ISL_19174530) is represented by strains that circulated between 2018 and 2023 in Asia (Fig. 3). The closest on the tree for the hemagglutinin gene were isolates obtained from China and Bangladesh between 2019–2023 (Fig. 3).

Fig. 3. Phylogenetic tree for the hemagglutinin gene for influenza A virus subtype H3N8 isolates.
Рис. 3. Филогенетическое дерево по гену гемагглютинина для изолятов вируса гриппа A субтипа H3N8.
The phylogenetic environment of the H3N8 isolate (EPI_ISL_ 19174467) also consists of Asian (China, Kazakhstan, Mongolia, Bangladesh) and European strains (Poland, Belgium, Germany, Austria) on the hemagglutinin gene. The closest isolates were strains that circulated in the territory of the Asian part of Russia during 2018‒2021 in wild waterfowl (Fig. 3).
Homology of sequenced isolate A/Pygoscelis papua/Fildes Peninsula, King George Island/31/2023 (H3N8) by the neuraminidase gene ranged from 96‒97% relative to isolates from wild waterfowl of Asian Russia A/common teal/Novosibirsk region/3556k/2020 (H3N8), Mongolia A/duck/Mongolia/WKU-42/2022 (H3N8) and European strains such as: A/duck/Chernogolovka/5897/2021(H3N8), A/Anas_platyrhynchos/Belgium/2987_0001/2021(H3N8), A/mallard/Germany-BB/2023AI07658/2023(H3N8). The maximum difference was observed between the studied isolate and A/duck/Bangladesh/19D2073/2023 (H3N8) strain (Fig. 4).

Fig. 4. Phylogenetic tree for the neuraminidase gene for influenza A virus subtype H3N8 isolates.
Рис. 4. Филогенетическое дерево по гену нейраминидазы для изолятов вируса гриппа A субтипа H3N8.
As a result of the study, the isolate A/Sterna paradisaea/Fildes Peninsula, King George Island/09/2023 (H3N8) was found to be phylogenetically closest to the Asian isolates A/mallard/Novosibirsk region/3314k/2020 (H3N8) and A/duck/Mongolia/29/2011 (H3N8) in terms of the neuraminidase gene. A significant difference (5%) was recorded between the nucleotide sequences of the studied isolate and the A/wild duck/Germany-NW/2023AI07895/2023 (H3N8) strain (Fig. 4). In general, the phylogeny of the studied isolates from Antarctica has approximately a similar trend with respect to the represented genes, being the contribution of both European and Asian strains to the reassortment, with all geographical locations united by territorial proximity to each other, such as the Netherlands, Belgium and Germany. Also, the results of the analysis showed that the studied isolates have a rather large genetic distance relative to the strains circulating in North America.
The eighth segment of influenza A virus RNA consists of 890 nucleotides and encodes 2 nonstructural proteins, NS1 and NS2. In 1980, C. Scholtissek and V. Von Hoyningen-Huene [27] determined that influenza A viruses can be divided into 2 groups (group 1 and the more heterogeneous group 2) with respect to the homology of the sequence of their non-structural genes (NS). Within this group, the homology ranges from 94 to 100% or 75 and 100%, respectively, whereas between the two groups it is in the range of only 40%. It is now common to define these groups as allele A and allele B, with allele B being associated exclusively with virus circulating only in birds and allele A being associated with viruses that have a predisposition to circulate in mammals. Also, the NS gene of most highly pathogenic H5N1 avian influenza viruses isolated in humans since 1997 is associated with the A allele [28].
The H3N8 subtype samples identified in this study belong to the Eurasian genetic lineage, with the NS gene belonging to the B allele. When comparing the nucleotide sequences of the studied isolates with strain A/Goose/Guangdong/1/1996, the level of homology was 92–93%, and with strain A/Mallard/Sweden/2724/2006 – 67%, respectively. The results of phylogenetic analysis on the NS gene of isolate A/Pygoscelis papua/ Fildes Peninsula, King George Island/31/2023 (H3N8) and isolate A/Sterna paradisaea/Fildes Peninsula, King George Island/09/2023 (H3N8) showed their significant relatedness (Fig. 5). The phylogenetic environment is represented mainly by specimens found in Belgium, the Netherlands and the European part of Russia. The maximum percentage of homology was observed between the presented isolates and strain A/Anas_platyrhynchos/Belgium/2214_0001/2021 (H3N8). The greatest difference in the NS gene was relative to strain A/aquatic bird/Korea/KN-1/2004 (H3N8).

Fig. 5. Phylogenetic tree for the NS1 gene for influenza A virus isolates of subtypes H3N8 and H1N1.
Рис. 5. Филогенетическое дерево по гену NS1 для изолятов вируса гриппа A субтипов H3N8 и H1N1.
Genetic analysis of the NS gene of the investigated sample of subtype H1N1 shows its correspondence to the Eurasian lineage, allele A. When compared with the sample A/Mallard/Sweden/2724/2006, the percentage of homology in nucleotide sequences was 87%. Relative to A/Goose/Guangdong/1/1996, the level of homology was 69%. The phylogenetic environment is represented by specimens from Korea, Bangladesh, China, Mongolia and the Asian part of the Russian Federation. A/Bean goose/Korea/KNU-16/2022 was the closest (98% similarity) by the NS gene to the studied specimen.
The samples from Argentina, Australia, and the United States had the greatest genetic distance from the identified isolate. The results of phylogenetic analysis of the NS gene suggest that influenza virus strains are genetically quite diverse and contain their own unique nucleotide sequences, which is most likely related to the function of this gene in the genome as an interferon antagonist (Fig. 5).
Thus, the results of this study demonstrated that the identified isolates were most likely the result of reassortment between European (Belgium, Netherlands, Russia) and Asian (Mongolia, China, Bangladesh) strains. A significant contribution to the formation of reassortants was caused by influenza A isolates circulating in the period 2018–2023, in the territory of the Asian part of Russia in wild waterfowl; this may indirectly indicate multiple introductions of different pathogen subtypes from the territory of the Asian part of Russia to the territory of Antarctica, with genetic variants circulating at the current time in Europe and Asia in wild bird populations.
It is known that successful interspecific transmission requires changes in the hemagglutinin properties of avian influenza viruses. It has been shown that these changes are associated with amino acid substitutions in the receptor-binding site of hemagglutinin [29].
The following amino acid substitutions associated with interspecies transmission of influenza virus hemagglutinin H1 have been found in the hemagglutinin H1, as evidenced by the literature: S138A, E190D, and G225D. For hemagglutinin H3, interspecies transmission was associated with Q226L and G228S mutations [30].
Discussion
Therefore, we analyzed amino acid sequences for the presence of such mutations in the identified H1N1 isolate. The analysis revealed amino acid substitutions S138Y and E190T, while there were no changes in position 225 corresponding to glycine.
For the A/Pygoscelis papua/Fildes Peninsula, King George Island/31/2023 (H3N8) isolate, the amino acid substitutions identified were proline at position 226, Q226P and tyrosine at 228 (G228T). Correspondingly for the isolate A/Sterna paradisaea/Fildes Peninsula, King George Island/09/2023 (H3N8): proline at position 226, Q226P and histidine at 228, G228H.
Thus, we can conclude that non-synonymous mutations have accumulated in the genomes of Antarctic isolates, but the impact of the corresponding amino acid substitutions on the biological properties of the viruses remains unexplored at present.
In general, the evolution of influenza viruses has a complex and dynamic character due to the peculiarities of genome organization and active circulation among a large number of wild waterfowl. The genomic diversity of zoonotic influenza viruses, which have already caused human infections, calls for strengthened surveillance of influenza viruses both among humans and in wildlife.
Ecosystems of coastal Antarctica are the most important geographical location from the point of view of organization of systematic monitoring studies. It should be noted that some species of Antarctic birds are capable of intercontinental migrations (for example, polar terns annually migrate from the Antarctic to the Russian Arctic). This fact actualizes the question of the role of long-distance migrants as vectors for the transfer of zoonotic infections, including influenza A.
Conclusion
The obtained data indicate the circulation in the Antarctic territory of influenza A viruses of subtypes H1N1 and H3N8 with unique genomic characteristics, which, in particular, is manifested by the spectrum of mutations associated with their potential interspecies transmission. The natural environment of Antarctica, despite extreme climatic conditions, is capable of forming possible zones of formation and evolution of viruses, in particular the influenza A virus. In our opinion, the study of genetic diversity of influenza A viruses circulating in the polar regions of the Earth and identification of inherent conditions for the emergence of new genetic variants and virus species has an important theoretical significance from the scientific point of view and is certainly an urgent task for the development of measures to prevent biological threats.
Funding. Obtaining biological material from Arctic terns (Sterna paradisaea) was carried out as part of research on the microbiota of Arctic bird species, supported by the Russian Science Foundation grant No. 23-25-00128 «Antibiotic-resistant bacteria associated with Arctic ornithogenic ecosystems: genetic characteristics and epidemic potential».
Acknowledgement. The authors express their gratitude to the leadership of the Russian Antarctic Expedition for assistance in conducting field studies. The authors are grateful to all researchers who participated in different stages of this work, especially to the staff of the Laboratory of Molecular Virology of the A. A. Smorodintsev Research Institute of Influenza for methodological support, as well as to V.L. Semin, Institute of Oceanology of the Russian Academy of Sciences.
Conflict of interest. The authors declare no apparent or potential conflicts of interest related to the publication of this article.
About the authors
Olesia V. Ohlopkova
Central Research Institute of Epidemiology, Rospotrebnadzor; Federal Research Center for Fundamental and Translational Medicine
Author for correspondence.
Email: ohlopkova.lesia@yandex.ru
ORCID iD: 0000-0002-8214-7828
PhD in Biology, Senior Researcher of the Laboratory of Experimental Pharmacology, Virology Research Institute
Russian Federation, 111123, Moscow; 630060, NovosibirskArtemy E. Goncharov
Institute of Experimental Medicine; Northwestern State Medical University named after I.I. Mechnikov
Email: phage1@yandex.ru
ORCID iD: 0000-0002-5206-6656
Doctor of Medical Sciences, Head of the Laboratory of Functional Genomics and Proteomics of Microorganisms; Professor of the Department of Epidemiology, Parasitology and Disinfection
Russian Federation, 197022, St. Petersburg; 191015, St. PetersburgBatyrbek I. Aslanov
Northwestern State Medical University named after I.I. Mechnikov
Email: Batyrbek.Aslanov@szgmu.ru
ORCID iD: 0000-0002-6890-8096
Doctor of Medical Sciences, Professor, and Head of the Department of Epidemiology, Parasitology and Disinfectology
Russian Federation, 191015, St. PetersburgArtem V. Fadeev
A.A. Smorodintsev Research Institute of Influenza, Ministry of Health of the Russian Federation
Email: artem.fadeev@influenza.spb.ru
ORCID iD: 0000-0003-3558-3261
Senior Researcher, Laboratory of Molecular Virology
Russian Federation, 197376, St. PetersburgYuri N. Davidyuk
Federal State Educational Institution of Higher Education «Kazan (Volga Region) Federal University»
Email: JNDavidjuk@kpfu.ru
ORCID iD: 0000-0002-4409-2942
PhD in Biology, Senior Researcher at the Institute of Fundamental Medicine and Biology
Russian Federation, 420008, KazanAlexey D. Moshkin
Federal Research Center for Fundamental and Translational Medicine
Email: alex.moshkin727@gmail.com
ORCID iD: 0000-0002-1182-8247
researcher at the Research Institute of Virology
Russian Federation, 630060, NovosibirskKristina A. Stolbunova
Federal Research Center for Fundamental and Translational Medicine
Email: kristina.sunwo@yandex.ru
ORCID iD: 0000-0003-3376-945X
researcher, Research Institute of Virology
Russian Federation, 630060, NovosibirskMarina A. Stepanyuk
Federal Research Center for Fundamental and Translational Medicine
Email: stepanunya1996@gmail.com
ORCID iD: 0009-0002-2658-7746
researcher, Research Institute of Virology
Russian Federation, 630060, NovosibirskIvan A. Sobolev
Federal Research Center for Fundamental and Translational Medicine
Email: sobolev_i@centercem.ru
ORCID iD: 0000-0002-4561-6517
PhD in Biology, Head of the Laboratory of Genomics and Evolution of Viruses, Research Institute of Virology
Russian Federation, 630060, NovosibirskMarina A. Tyumentseva
Central Research Institute of Epidemiology, Rospotrebnadzor
Email: tyumantseva@cmd.ru
ORCID iD: 0000-0002-3145-3702
PhD in Biology, Head of the Laboratory of Genomic Editing of the Department of Molecular Diagnostics and Epidemiology
Russian Federation, 111123, MoscowAlexander I. Tyumentsev
Central Research Institute of Epidemiology, Rospotrebnadzor
Email: tyumantsev@cmd.ru
ORCID iD: 0000-0003-0537-2586
PhD in Biology, Head of the Laboratory of Experimental Pharmacology, Department of Molecular Diagnostics and Epidemiology
Russian Federation, 111123, MoscowAlexander M. Shestopalov
Federal Research Center for Fundamental and Translational Medicine
Email: amshestopalov@frcftm.ru
ORCID iD: 0000-0002-9734-0620
Doctor of Biological Sciences, Professor, Honored Worker of Science of the Russian Federation, Director of the Research Institute of Virology
Russian Federation, 630060, NovosibirskVasily G. Akimkin
Central Research Institute of Epidemiology, Rospotrebnadzor
Email: akinkin@pcr.ms
ORCID iD: 0000-0003-4228-9044
Doctor of Medical Sciences, Academician of the Russian Academy of Sciences, Professor, Honored Doctor of the Russian Federation, Laureate of the Prize of the Government of the Russian Federation in the field of science and technology, Director
Russian Federation, 111123, MoscowReferences
- Bouvier N.M., Palese P. The biology of influenza viruses. Vaccine. 2008; 26(Suppl. 4): D49–53. https://doi.org/10.1016/j.vaccine.2008.07.039
- Fries A.C., Nolting J.M., Bowman A.S., Lin X., Halpin R.A., Wester E., et al. Spread and persistence of influenza A viruses in waterfowl hosts in the North American Mississippi migratory flyway. J. Virol. 2015; 89(10): 5371–81. https://doi.org/10.1128/JVI.03249-14
- Lvov D.K., Zhdanov V.M. Circulation of influenza virus genes in the biosphere. In: Zhdanov V.M., ed. Soviet Medical Reviews. Section E: Virology Reviews. Volume 1. Harwood Academic Publ. GmbH; 1984: 129–52.
- Lvov D.K. Circulation of Influenza Virus in Natural Biocenosis. In: Kurstak E., Maramorosh K., eds. Viruses and Enviroment. Academic Press; 1978: 351–80.
- Lvov D.K. Influenza A virus – a sum of populations with a common protected gene pool. In: Zhdanov V.M., ed. Soviet Medical Reviews. Section E: Virology Reviews. Volume 2. Harwood Academic Publ. GmbH; 1984: 15–37.
- Lvov D.K., Kaverin N.V. Avian influenza in Northern Eurasia. In: Klenk H.D., Matrosovich M.N., eds. Avian Influenza. Volume 27. Basel: Karger; 2008: 41–58.
- Lvov D.K., Ilyichev V.D., ed. Bird Migration and Transmission of Infectious Agents [Migratsiya ptits i perenos vozbuditelei infektsii]. Moscow: Nauka; 1979. (in Russian)
- Walker P.J., Siddell S.G., Lefkowitz E.J., Mushegian A.R., Adriaenssens E.M., Alfenas-Zerbini P., et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses. Arch. Virol. 2022; 167(11): 2429–40. https://doi.org/10.1007/s00705-022-05516-5
- Gass J.D. Jr., Dusek R.J., Hall J.S., Hallgrimsson G.T., Halldórsson H.P., Vignisson S.R., et al. Global dissemination of influenza A virus is driven by wild bird migration through arctic and subarctic zones. Mol. Ecol. 2023; 32(1): 198–213. https://doi.org/10.1111/mec.16738
- Wille M., Holmes E.C. The ecology and evolution of influenza viruses. Cold. Spring Harb. Perspect. Med. 2020; 10(7): a038489. https://doi.org/10.1101/cshperspect.a038489
- Cowling B.J., Ip D.K., Fang V.J., Suntarattiwong P., Olsen S.J., Levy J., et al. Aerosol transmission is an important mode of influenza A virus spread. Nat. Commun. 2013; 4: 1935. https://doi.org/10.1038/ncomms2922
- Barratclough A., Ferguson S.H., Lydersen C., Thomas P.O., Kovacs K.M. A review of circumpolar arctic marine mammal health – a call to action in a time of rapid environmental change. Pathogens. 2023; 12(7): 937. https://doi.org/10.3390/pathogens12070937
- Lvov D.K. Ecology of viruses. In: Lvov D.K., ed. A Guide to Virology. Viruses and Viral Infections of Humans and Animals [Rukovodstvo po virusologii. Virusy i virusnye infektsii cheloveka i zhivotnykh]. Moscow: MIA; 2013: 66–86. (in Russian)
- Lvov D.K., Zdanov V.M., Sazonov A.A., Braude N.A., Vladimirtceva E.A., Agafonova L.V., et al. Comparison of influenza viruses isolated from man and from whales. Bull World Health Organ. 1978; 56(6): 923–30.
- Nielsen O., Clavijo A., Boughen J.A. Serologic evidence of influenza A infection in marine mammals of Arctic Canada. J. Wildl. Dis. 2001; 37(4): 820–25. https://doi.org/10.7589/0090-3558-37.4.820
- Celis J.E., Espejo W., Barra R., Gonzalez-Acuña D., Gonzalez F., Jara S. Assessment of trace metals in droppings of Adélie penguins (Pygoscelis Adeliae) from different locations of the Antarctic peninsula area. Adv. Polar Sci. 2015; 26(1): 1–7. https://doi.org/10.13679/j.advps.2015.1.00001
- Hahn S., Peter H.U., Quillfeldt P., Reinhardt K. The birds of the Potter Peninsula, King George Island, South Shetland Islands, Antarctica, 1965-1998. Marine Ornithology. 1998; 26: 1–6.
- Wille M., Aban M., Wang J., Moore N., Shan S., Marshall J., et al. Antarctic penguins as reservoirs of diversity for avian avulaviruses. J. Virol. 2019; 93(11): e00271-19. https://doi.org/10.1128/JVI.00271-19
- Siniff D.B., Garrott R.A., Rotella J.J., Fraser W.R., Ainley D.G. Opinion: Projecting the effects of environmental change on Antarctic seals. Antarct. Sci. 2008; 20(5): 425–35. doi: 10.1017/S0954102008001351
- Shao W., Li X., Goraya M.U., Wang S., Chen J.L. Evolution of influenza A virus by mutation and re-assortment. Int. J. Mol. Sci. 2017; 18(8): 1650. https://doi.org/10.3390/ijms18081650
- Thorsson E., Zohari S., Roos A., Banihashem F., Bröjer C., Neimanis A. Highly pathogenic avian influenza A(H5N1) virus in a Harbor porpoise, Sweden. Emerg. Infect. Dis. 2023; 29(4): 852–5. https://doi.org/10.3201/eid2904.221426
- Markin A., Wagle S., Grover S., Vincent Baker A.L., Eulenstein O., Anderson T.K. PARNAS: objectively selecting the most representative taxa on a phylogeny. Syst. Biol. 2023; 72(5): 1052–63. https://doi.org/10.1093/sysbio/syad028
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014; 30(9): 1312–3. https://doi.org/10.1093/bioinformatics/btu033
- Short K.R., Richard M., Verhagen J.H., van Riel D., Schrauwen E.J., van den Brand J.M., et al. One health, multiple challenges: The inter-species transmission of influenza A virus. One Health. 2015; 1: 1–13. https://doi.org/10.1016/j.onehlt.2015.03.001
- Al Hajjar S., McIntosh K. The first influenza pandemic of the 21st century. Ann. Saudi Med. 2010; 30(1): 1–10. https://doi.org/10.4103/0256-4947.59365
- Yu J., Yao Q., Liu J., Zhou Y., Huo M., Ge Y. Concern regarding H3-subtype avian influenza virus. Front. Microbiol. 2023; 14: 1327470. https://doi.org/10.3389/fmicb.2023.1327470
- Scholtissek C., von Hoyningen-Huene V. Genetic relatedness of the gene which codes for the nonstructural (NS) protein of different influenza A strains. Virology. 1980; 102(1): 13–20. https://doi.org/10.1016/0042-6822(80)90065-3
- Jahangir A., Ruenphet S., Sultana N., Shoham D., Takehara K. Genetic analysis of avian influenza viruses: cocirculation of avian influenza viruses with allele A and B nonstructural gene in Northern Pintail (Anas acuta) ducks wintering in Japan. Influenza Res. Treat. 2012; 2012: 847505. https://doi.org/10.1155/2012/847505
- Herfst S., Zhang J., Richard M., McBride R., Lexmond P., Bestebroer T.M., et al. Hemagglutinin traits determine transmission of avian A/H10N7 influenza virus between mammals. Cell Host Microbe. 2020; 28(4): 602–13.e7. https://doi.org/10.1016/j.chom.2020.08.011
- Shi Y., Wu Y., Zhang W., Qi J., Gao G.F. Enabling the ‘host jump’: structural determinants of receptor-binding specificity in influenza A viruses. Nat. Rev. Microbiol. 2014; 12(12): 822–31. https://doi.org/10.1038/nrmicro3362
Supplementary files

