



The Molecular and Biological Patterns Underlying Sustained SARS-CoV-2 Circulation in the Human Population
- Authors: Kustova D.D.1,2, Pochtovyi A.A.1,2,3, Shpakova O.G.4, Shtinova I.A.4, Kuznetsova N.A.1, Kleimenov D.A.1, Komarov A.G.4, Gushchin V.A.1,2,3
-
Affiliations:
- National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation
- Federal State Budgetary Educational Institution of Higher Education Lomonosov Moscow State University
- I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University)
- Moscow Healthcare Department
- Issue: Vol 69, No 4 (2024)
- Pages: 329-340
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16651
- DOI: https://doi.org/10.36233/0507-4088-242
- EDN: https://elibrary.ru/uxnluj
- ID: 16651

Cite item

Abstract
Introduction. For four years, SARS-CoV-2, the etiological agent of COVID-19, has been circulating among humans. By the end of the second year, an absence of immunologically naive individuals was observed, attributable to extensive immunization efforts and natural viral exposure. This study focuses on delineating the molecular and biological patterns that facilitate the persistence of SARS-CoV-2, thereby informing predictions on the epidemiological trajectory of COVID-19 toward refining pandemic countermeasures.
The aim of this study was to describe the molecular biological patterns identified that contribute to the persistence of the virus in the human population.
Materials and methods. For over three years since the beginning of the COVID-19 pandemic, molecular genetic monitoring of SARS-CoV-2 has been conducted, which included the collection of nasopharyngeal swabs from infected individuals, assessment of viral load, and subsequent whole-genome sequencing.
Results. We discerned dominant genetic lineages correlated with rising disease incidence. We scrutinized amino acid substitutions across SARS-CoV-2 proteins and quantified viral loads in swab samples from patients with emerging COVID-19 variants. Our findings suggest a model of viral persistence characterized by 1) periodic serotype shifts causing substantial diminutions in serum virus-neutralizing activity (> 10-fold), 2) serotype-specific accrual of point mutations in the receptor-binding domain (RBD) to modestly circumvent neutralizing antibodies and enhance receptor affinity, and 3) a gradually increasing amount of virus being shed in mucosal surfaces within a single serotype.
Conclusion. This model aptly accounts for the dynamics of COVID-19 incidence in Moscow. For a comprehensive understanding of these dynamics, acquiring population-level data on immune tension and antibody neutralization relative to genetic lineage compositions is essential.
Keywords
Full Text
Introduction
The COVID-19 pandemic was caused by a novel SARS-CoV-2 virus discovered in late 2019 and has caused more than 774 million cases and more than 7 million deaths worldwide [1]. The observed evolution of SARS-CoV-2 has led to the independent emergence of several genetic lineages identified by the World Health Organization at certain periods as variants of concern (VOC) – Alpha, Beta, Gamma, Epsilon, Delta and Omicron [2], which have been associated with upsurges in morbidity and mortality in most countries. Despite the rapid development and introduction of effective prophylactic drugs following the first year of the pandemic [3–6], as the first VOCs emerged, reports of decreased vaccine efficacy began to emerge [7–9]. This became the core reason for the introduction of booster doses [10, 11]. In the case of the Omicron variant, efficacy decreased to such an extent that booster doses with a modified formulation had to be administered [12–15]. However, in the case of the relatively recent XBB variant, even with the use of an updated divalent booster followed by a monovalent vaccine containing the XBB antigen, a decrease in neutralization has been observed [8]. Furthermore, a large amount of information has now been accumulated on the impact of mutations characteristic of dominant variants on the efficacy of therapeutic agents including monoclonal antibodies and antiviral agents [16–18], which has resulted in the loss of efficacy of most monoclonal antibodies (mAbs) against topically circulating variants [19–21]. The emergence of a new virus variant was preceded by the emergence of mutations, which in turn affected changes in the transmissibility and/or pathogenicity of the virus, sensitivity to therapeutic drugs and ability to evade natural or vaccine-induced immunity. It is worth noting that indicators such as viral transmissibility and/or pathogenicity are inextricably linked to the magnitude of viral load. As discussed previously [22], host (role of vaccination or prior infection) and viral factors (SARS-CoV-2 variants) significantly influence viral load dynamics and therefore further influence viral transmission. Despite the accumulated data on the values of some mutations and associated phenotypic traits [22], the factors influencing the dynamics of the epidemiologic process in different periods remain incompletely understood.
This study is based on the results of molecular genetic monitoring of SARS-CoV-2 virus variants, some of which have been previously published [23]. In this study, we assessed both the dynamics of circulating variants and the potential impact of mutations and viral load as the main factors contributing to changes in the profile of circulating lineages, which may be reflected in the observed transition: «increase in mutations ‒ increase in viral load ‒ emergence of a new serotype».
Materials and methods
During the period of Delta variant emergence, nasopharyngeal swabs were collected regularly from different groups of volunteers, including those who were initially infected or vaccinated. Written consent was obtained from all patients in accordance with the order of the Russian Ministry of Health of July 21, 2015, No. 474n «On the procedure for giving informed voluntary consent to medical care within the framework of clinical approbation of methods of prevention, diagnosis, treatment, and rehabilitation, the forms of informed voluntary consent to medical care within the framework of clinical approbation of methods of prevention, diagnosis, treatment, and rehabilitation, and refusal of medical care within the framework of clinical approbation of methods of prevention, diagnosis, treatment, and rehabilitation». All samples were depersonalized before they were received by the research team. The study was approved by the local ethical committee of the N.F. Gamaleya NRCEM (protocol No. 14, September 29, 2021).
Sample collection and viral load testing were performed in two laboratories (hereinafter referred to as Laboratory 1 and Laboratory 2). During the circulation period of Wuhan, Delta and Omicron variants (BA.1.X and BA.2.X) (Laboratory 1), nasopharyngeal swabs were collected in virus transport medium (G00155, GEM, Russia). Total RNA was isolated using the QIAamp Viral RNA Mini Kit (Qiagen, Germany) and a kit for isolation of total RNA from animal and bacterial cells, smears and viruses on columns (RU-250, Biolabmix, Novosibirsk, Russia). Quantitative reverse transcription polymerase chain reaction (PCR) was performed using the SARS-CoV-2 FRT RT-PCR kit (EA-128, N.F. Gamaleya NRCEM, Moscow, Russia). Samples with Ct values < 30 were selected for full-genome sequencing.
In Laboratory 2, nasopharyngeal swabs were collected in a virus transport medium (physiological solution or XK-PCR30 transport medium (Jiangsu Xinkang Medical Instrumet Co., Ltd., China)) or GEM transport medium (GEM, Moscow, Russia). During the circulation period of Omicron BA.1.X, BA.2.X, BA.5.X variants, quantitative reverse transcription PCR was performed using the AmpliPrime SARS-CoV-2 DUO kit (NextBio, Moscow, Russia) according to the manufacturer’s instructions. During the circulation period of Omicron BA.5.X, CL.X, XBB.1.X and XBB.1.9.X variants, quantitative reverse transcription PCR was performed using CoV-2 test (TestGen, Ulyanovsk, Russia) according to the manufacturer’s instructions.
Further whole genome sequencing was performed using Ion Torrent (Thermo Fisher Scientific, USA), Illumina (Illumina, USA), and Oxford Nanopore (Oxford, UK) technologies. The sample preparation and analysis procedures have been described in detail previously [21, 23]. The whole genome sequences obtained in this study were uploaded to the GISAID database under the following numbers: EPI_ISL_1710849-1710866, EPI_ISL_2296111-2296286, EPI_ISL_2296288-2296379, EPI_ISL_4572812, EPI_ISL_5334362-5334371, EPI_ISL_5334374-5334389, EPI_ISL_7211325-7211326, EPI_ISL_7263932-7263933, EPI_ISL_9230058-9230062, EPI_ISL_9230064-9230100, EPI_ISL_10627062, EPI_ISL_11864996-11865125, EPI_ISL_11872910, EPI_ISL_421275, EPI_ISL_454732, EPI_ISL_470896-470904, EPI_ISL_572398, EPI_ISL_872628-872643, EPI_ISL_875515, EPI_ISL_1015362, EPI_ISL_1708507-1708509, EPI_ISL_12225322, EPI_ISL_12748381-12748382, EPI_ISL_13431664-13431687, EPI_ISL_14217225-14217226, EPI_ISL_15327072-15327075, EPI_ISL_15858138-15859137, EPI_ISL_15860713-15860737, EPI_ISL_15860739-15860839, EPI_ISL_15860841-15860991, EPI_ISL_15860993-15861048, EPI_ISL_15862338-15863336, EPI_ISL_15863677-15864655, EPI_ISL_15864802-15865776, EPI_ISL_15865821-15866801, EPI_ISL_15867150-15868141, EPI_ISL_15868158-15869145, EPI_ISL_15869218-15870209, EPI_ISL_15871156-15872140, EPI_ISL_15872157-15873150, EPI_ISL_15873159-15874146, EPI_ISL_15874159-15875146, EPI_ISL_15875638-15876623, EPI_ISL_15876640-15877626, EPI_ISL_15879747-15880730, EPI_ISL_15883551-15884536, EPI_ISL_15884833-15885823, EPI_ISL_15885995-15886980. The sequences obtained were also uploaded to the VGARus database.
The information on the circulation of genetic lineages used in this study was supplemented with data obtained from the GISAID database. The obtained data were filtered by the following parameters: Host == “Human”, `Sequence length` >= 27000, `Is complete?` == “TRUE” and Location == “Europe/Russia/Moscow/Moscow” or “Europe/Russia/Moscow” (query date September 18, 2023). Additionally, the retrieved sequences were also filtered by collection date (`Collection date` < “2023-06-01”), resulting in 16,541 records.
The genetic variant information for the remaining sequences was grouped as follows: Wuhan (B.1 + B.1.X), Alpha (B.1.1.7 + Q.X), Beta (B.1.351), Delta (B.1.617.X + AY.X), BA.1.X (B.1.1.529 + BA.1.X), BA.2.X, BA.5.X (BA.5.X + BF.X + BE.X), CL.X, XBB.1.X (without XBB.1.9.X and XBB.1.16.X), XBB.1.9.X, XBB.1.16.X (XBB.1.16.X + FU.X), and «Other XBB»/«Other XBB» (Other XBB lineages + EG.X). Furthermore, the genetic lineages B.1.1.317 and B.1.1.523 were added. The remaining lineages were combined into the «Other»/«Other» group. The obtained data were merged with metadata and visualized in R environment using the dplyr v. 1.1.1.1 [24] and ggplot2 v. 3.4.2 [25] packages.
The Kraskell–Wallis test was used to compare more than two independent groups. If the results were statistically significant, Dunn’s test was applied to determine the difference between groups. A comparison of PCR threshold cycle values was performed among samples where the same quantitative reverse transcription PCR kit was used. For statistical analysis, the ggstatsplot v. 0.12.1 [26] and R v. 4.2.2 packages were used.
Results
Dynamics of SARS-CoV-2 genetic variants in Moscow since the beginning of the COVID-19 pandemic
Since the initial spread of SARS-CoV-2 in Moscow up until February 2021, the predominant strains were characterized as Wuhan (B.1.X), including its diverse genetic sublineages, among which the circulation of a primarily Russia-specific genetic lineage, B.1.1.317, is notable. In winter 2021, the previously circulating variants began to be gradually replaced by the Alpha (B.1.1.7 + Q*) and B.1.1.523 lineages (also characteristic mainly of Russia), which were displaced by the Delta variant (B.1.617.2 + AY*) in May 2021 together with the next COVID-19 incidence increase. By the end of 2021, the Omicron variant (B.1.1.529) succeeded Delta, followed by sequential transition of the genetic lineages BA.1.X, BA.2.X, and BA.5.X. In October 2022, CL.X becomes one of the dominant variants, such a widespread distribution of which has not been reported elsewhere. Along with CL.X, a recombinant variant XBB began circulating in Moscow in November 2022, subsequently displacing the other SARS-CoV-2 genetic lineages (Fig. 1 a).

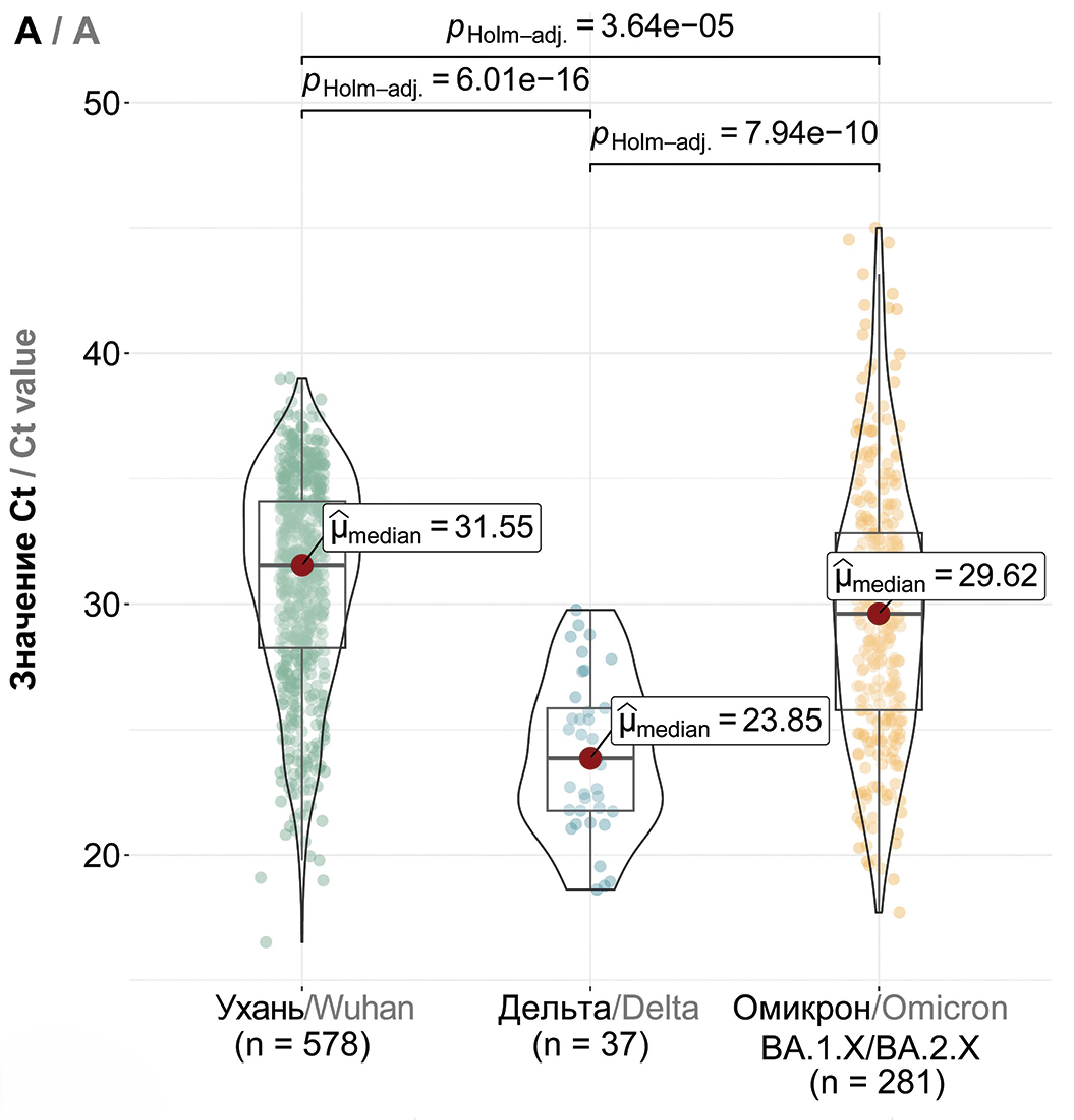
Fig. 1. Genetic variants of SARS-CoV-2 circulating in Moscow since the start of the COVID-19 pandemic. a – dynamics of genetic variants of SARS-CoV-2 circulating in Moscow. The left Y-axis indicates the proportion of genetic lineages, while the right axis represents the number of new COVID-19 cases per 100,000 population; b – changes in the number of mutations in the predominant circulating variants of SARS-CoV-2. The ordinate axis displays the mutation count. The periods (months) are indicated on the abscissa axis.
Рис. 1. Генетические варианты SARS-CoV-2, циркулировавшие в Москве с начала пандемии COVID-19. a – динамика генетических вариантов SARS-CoV-2. Слева по оси ординат показана доля генетических линий, справа – число новых случаев COVID-19 на 100 тыс. населения; б – изменение количества мутаций в основных циркулировавших вариантах SARS-CoV-2. По оси ординат показано количество мутаций. На оси абсцисс отражены периоды (месяцы).
Different variants of the virus, which became relatively widespread, had a characteristic profile of mutations.
Consideration of their total number in the S-protein and other genes showed that before the appearance of the Omicron variant there was a gradual increase in their number; the spread of the latter was characterized by a more than twofold increase in the number of nonsynonymous mutations in the SARS-CoV-2 genome (Fig. 1 b). Subsequently, substitutions also continued to appear in the viral genome, but the first Omicron variant, BA.1.X, characterized by 34 mutations in the S-protein, which is 3 more than in the subsequent BA.2.X, and comparable to BA.5.X, falls out of the general series. Despite this fact, the total number of mutations in the genome of the SARS-CoV-2 variant BA.1.X was the lowest among all other Omicron lines and amounted to 60, whereas the last variant considered, XBB.1.9.X, had about 80 amino acid substitutions, of which 41 were in the S-protein.
Mutation profile in the major genetic variants of SARS-CoV-2.
A detailed review of mutations in the selected variants revealed a characteristic mutation profile for the SARS-CoV-2 variants circulating in Moscow. In the RBD of the S-protein, the N501Y mutation was observed for Alpha, E484K and S494P for B.1.1.523. Delta was characterized by two amino acid substitutions (L452R and T478K) in the RBD of the S-protein, and 13 for BA.1.X, and the occurrence of the K417N, N440K, and G446S mutations in some genetic lineages related to this Omicron variant was also observed. The BA.2.X variant is notable for 16 mutations in the RBD, of which only 10 are common with BA.1.X; the dominating variants do not exhibit the R346K and S371L mutations present in BA1.X, and, furthermore, a series of additional substitutions appeared, including S371F, T376A, D405N, R408S, K417N, and N440K. The variants BA.5.X and CL.X had the L452R mutation characteristic of the Delta lineage and F486V, and all further circulating variants were also characterized by amino acid substitutions at position 486. From the end of 2022, the dominant genetic lineage in Moscow was XBB and its subvariants (Fig. 1) with a characteristic minimum of 22 amino acid mutations in RBD, of which 6 were not found in previously circulating variants (Fig. 2). In spring 2023, the main circulating variant was XBB.1.9.X, namely XBB.1.9.1, characterized by the F486P substitution (Fig. 2).

Fig. 2. Mutations in the RBD of the spike protein of the predominant SARS-CoV-2 variants. The Y-axis represents SARS-CoV-2 variants, and the X-axis represents amino acid substitutions. Zero values reflect the proportion of mutations tending to zero; empty cells represent the absence of mutations at a specific position.
Рис. 2. Мутации в RBD Spike-белка основных доминировавших вариантов SARS-CoV-2. По оси ординат отражены варианты SARS-CoV-2, по оси абсцисс – аминокислотные замены. Нулевые значения отражают долю мутаций, стремящуюся к нулю; пустые ячейки – отсутствие мутаций в данной позиции.
A wide variety of amino acid substitutions was observed in the N-terminal domain (NTD) of the S-protein. The genetic lineage B.1.1.523 was characterized by the presence of the F306L mutation. Genetic lineages belonging to the Delta variant had mutations that were not subsequently found in other variants: T19R, E156G, F157del, R158del, and one G142D mutation characteristic of all Omicron lineage variants except BA.1.X. Overall, most BA.1.X mutations were uncharacteristic of subsequent Omicron variants with the exception of deletions 69‒70, which were also present in BA.5.X and CL.X. CL.X, compared to BA.5.X and other Omicron variants, possessed a K150E amino acid substitution; otherwise, variants BA.2.X, BA.5.X, and CL.X had a similar mutation profile. The XBB variants were characterized by V83A, H146Q, Q183E, and V213E mutations not previously seen in other genetic lineages. All variants had a D614G substitution in the C-terminal domain (CTD), which is known to be fixed before the SARS-CoV-2 genetic lineages were divided into VOCs [27]. For Alpha, the largest number of amino acid mutations was observed in the CTD region: P681H, T716I, S982A, and D1118H, not including D614G. B.1.1.523 was characterized by the presence of D839V and T1027I, not further observed in other variants. Delta in CTD had only 2 substitutions besides D614G: P681R near the furin site and D950N. Further, BA.1.X had the highest number of mutations in this region compared to the other Omicron variants: in addition to the characteristic H655Y, N679K, N681H, N764K, D796Y, Q954H, N969K, the first Omicron variant had additional T547K, N856K, and L981F. Overall, little further evolution of the Omicron NTD was observed, but T883I substitution was observed in CL.X and XBB.1.X in a small percentage of cases (29 and 12, respectively) (Fig. S3 a).
The frequency of mutations in non-structural genes was much lower than in the S-protein. The Delta variant was characterized by about 16 mutations in nonstructural genes, and mutations T492I in NSP4 and P323L in RdRp (NSP12) were also characteristic of all subsequent Omicron variants, and G671S in the same gene was characteristic of XBB genetic lineages. The remaining mutations, however, were not further encountered. The first Omicron variant, BA.1.X, had amino acid substitutions NSP3_K38R, NSP3_L1266I, NSP3_A1892T, NSP6_I189V and deletions NSP3_S1265del and NSP6_L105del, which were not found in Omicron variants, as well as a number of other mutations characteristic of Omicron: NS9b_P10S, NS9b_E27-29del, NSP4_T492I, NSP5_P132H, NSP6_S106-107del, and NSP14_I42V. All subsequent Omicron variants compared to BA.1.X possessed NS3a_T223I, NSP1_S135R, NSP3_T24I, NSP3_G489S, NSP4_L264F, NSP4_T327I, NSP6_F108del, NSP13_R392C, and NSP15_T112I mutations. Distinctive features of BA.2.X and XBB were amino acid substitutions in NS6 ‒ D61L and NSP4 ‒ L438F. BA.5.X was characterized by additional mutations NS9b_D16G and NSP13_T127N, which were not found in other variants. CL.X, although being a sublineage of BA.5.X, did not possess these substitutions, but was characterized by the presence of NS8_F41C and NSP13_N102S. XBB variants, in addition to the above mutations, possessed a stop codon at the 8th position of NS8 and NSP1_K47R, while XBB.1.9.X possessed NS9b_I5T, NSP3_G1001S, NSP9_T35I and, in a small percentage of cases, NSP2_D449E (Fig. S3 c).
Regarding other structural genes of SARS-CoV-2, the Delta variant was not characterized by mutations in the E-protein, but substitutions in the matrix protein (M_I82T) and nucleocapsid protein (N_D63G, N_R203M, N_G215C, N_D377Y) were observed, and, as with most other amino acid substitutions, they were not found in Omicron. All Omicron variants circulating in Moscow were characterized by the following mutations: E_T9I, M_Q19E, M_A63T, N_P13L, N_E31-33del, N_R203K, N_G204R, but some genetic lineages had their own peculiarities. For example, BA.1.X carried an additional M_D3G mutation to those listed, the BA.2.X variant and all other Omicron variants considered were N_S413R, BA.5.X and CL.X carried M_D3N. CL.X also possessed the N_A218S substitution, and all XBBs possessed the E_T11A substitution (Fig. S3 b).
Viral load dynamics of the main circulating variants
Comparative analysis of viral load (equivalent - PCR threshold cycle, Ct) showed that among the Wuhan (B.1.X), Delta (B.1.617.2 + AY.X) and the first Omicron variants (BA.1/BA.2), the highest viral load was characteristic of the Delta variant with a median value of 23.85 (p < 0.0001, Fig. 3 a). Thus, an increase in viral load was observed when the Wuhan variant was replaced by genetic lineages belonging to the Delta variant (within the same “serotype”). The first Omicron variants were characterized by a reduced viral load compared to Delta (median 29.62, p < 0.0001), and further there was also a statistically significant small increase in Ct values for variants BA.2.X and BA.5.X compared to BA.1.X (Fig. 3 b). No statistically significant differences were found between BA.2.X and BA.5.X. The CL.X variant was characterized by a reduced Ct value compared to BA.5.X with median values of 35.13 and 27.27, respectively (p < 0.0001, Fig. 3 c). Furthermore, CL virus variants with the additional T883I mutation showed a higher viral load compared to variants without this mutation (p = 0.001, Fig. S1), but the viral load of XBB.1.X was not affected by the presence of this mutation (Fig. S1). There was a trend towards higher viral load in successive variants BA.5.X, CL.X, XBB.1.X and XBB.1.9.X, where the highest viral load was characterized for XBB.1.9.X (p < 0.01, Fig. 3 c).

Fig. 3. Comparative analysis of viral load in predominant SARS-CoV-2 variants in Moscow. a – viral load in Wuhan (B.1.X), Delta (B.1.617.2 + AY.X), and Omicron (BA.1/BA.2.X) variants; b – viral load in Omicron BA.1.X, BA.2.X, and BA.5.X variants; c – viral load in BA.5.X, CL.X, XBB.1.X, and XBB.1.9.X variants. The Y-axis denotes Ct values, while the X-axis represents SARS-CoV-2 variants and the number of samples.
Рис. 3. Сравнение вирусной нагрузки основных доминировавших в Москве вариантов SARS-CoV-2. a – вирусная нагрузка вариантов Ухань (B.1.X), Дельта (B.1.617.2 + AY.X) и Омикрон (BA.1/BA.2.X); б – вирусная нагрузка вариантов Омикрон BA.1.X, BA.2.X и BA.5.X; в – вирусная нагрузка вариантов BA.5.X, CL.X, XBB.1.X и XBB.1.9.X. По оси ординат отражены значения Ct, по оси абсцисс – варианты SARS-CoV-2 и количество образцов.
Discussion
During more than 4 years of the COVID-19 pandemic, an unprecedented amount of data, primarily genomic data, was generated that is being used both to understand evolution and to improve prophylactic and therapeutic agents. During this period, virus variants with different phenotypic characteristics, including infectivity, disease severity, and immune evasion, have emerged [28]. A comprehensive analysis of the obtained data on the pathogenesis of SARS-CoV-2 allows us to understand the underlying mechanisms (drivers) of evolution and to recognize the processes that generate this diversity to potentially predict possible future variants of the virus. As the main drivers of the ongoing pandemic, we considered two key aspects – viral load and the profile of mutations in the virus genome, which generally characterizes the observed transition: «increase in mutations – increase in viral load – emergence of a new serotype».
From the pandemic’s onset in 2020 until the early summer of 2023, Moscow witnessed the succession of several genetic variants of SARS-CoV-2, namely B.1.X, Delta (B.1.617.2 + AY.X), and Omicron. Within each of these, certain genetic lineages were dominant at the respective time periods: in 2020. – B.1.1.317, in 2021 – B.1.1.523, Alpha (B.1.1.7 + Q.4) and AY.122 [29], in 2022 – lineages BA.1.X, BA.2.X, BA.5.X, CL.X, in 2023 – recombinant XBB with the respective sublines XBB.1.X and XBB.1.9.X (Fig. 1 а). A consistent feature was a consistent increase in the number of nonsynonymous mutations in the genome of circulating SARS-CoV-2 variants, about half of which were in the S-protein (Fig. 1 b). Subsequent detailed consideration of the molecular-genetic properties of circulating variants of SARS-CoV-2, including the profile of mutations in the SARS-CoV-2 genome and viral load, allowed us to identify certain patterns that could account for the continued spread of new variants of the COVID-19 pathogen and the newly arising rises in morbidity and hospitalizations in Russia and worldwide. Thus, it was found that when switching from variants B.1.X to Delta (B.1.618.2 + AY.X), there was a decrease in viral load by 7.7 PCR cycles, and this phenomenon was previously also observed in other studies [30, 31]. This variant was also characterized by several mutations, including those in the receptor-binding domain of the Spike protein (Fig. 2, Fig. S3), which could potentially account for its increased viral load. In previous studies, the presence of the L452R substitution in the S-protein has been shown to correlate with evasion of neutralizing antibodies [32], increased affinity for the ACE2 receptor, and increased spike stability and viral infectivity, thereby contributing to increased viral replication [33]. Furthermore, mutations E156G, T478K and D614G also increased infectivity and viral affinity for the receptor, while other substitutions such as P681R and D950N promoted higher propagation rate due to more efficient cleavage of S1/S2 at the furin site [34, 35]. In addition to structural proteins, other mutations could also influence the spread of the Delta variant and increase its viral load, respectively. For example, the T492I mutation in NSP4 was shown to have a positive effect on virus replication [36]. Virus variants with the P323L amino acid substitution in the RdRp gene in vitro had a selective advantage over variants without it, and, in addition, the presence of L323 and S671 was associated with more efficient replication at reduced temperature in the upper respiratory tract [37]. Thus, additional mutations characteristic of Delta could directly result in a high viral load, which could determine the faster spread of this variant compared to the previously circulating variants [23, 38, 39].
The first Omicron BA.1/BA.2.X variants, which replaced the Delta variant, were characterized, on the contrary, by a reduced viral load with a higher rate of spread [40]. It is worth noting that Omicron is phylogenetically divergent from previously circulating variants and possesses more than 15 substitutions in the RBD alone (Fig. 2), not counting mutations in other genes (Fig. S3). In addition, this variant is serologically distinct from previously circulating variants, as evidenced by several studies. Whereas vaccination efficacy against Delta remained high [41] and most monoclonal antibodies neutralized the virus [42], in the case of Omicron, a decrease in vaccination efficacy and sensitivity to therapeutic and prophylactic drugs was observed [7, 8]. Thus, it can be assumed that Omicron spread actively at the first stages not due to increased viral load, but due to the fact that the immune system had not encountered such a virus serotype before [43]. Furthermore, mutations in nonstructural proteins (e.g., 203K/204R in the N-protein and ΔSGF/ΔLSG in NSP6) and S-protein (N501Y and H655Y) acquired during the evolution of the Omicron variant may also have contributed to its widespread spread due to its high replication rate and more efficient virus transmission [44–47]. Furthermore, the dramatic increase in the number of mutations in the RBD may be a consequence of their mutual influence, where the appearance of one substitution entailed the appearance of another, as in the case of Q498R and N501Y, which also affected ACE2 binding affinity and infectivity of the virus, respectively [48]. Since the second half of 2022, BA.5.X has become the dominant Omicron variant in Moscow, thereby replacing the previously circulating BA.2.X (Fig. 1 a). Comparative analysis showed that the viral load of genetic lineages BA.2.X and BA.5.X was statistically significantly different from BA.1.X with median Ct values of 25.1, 25.0, and 24.3, respectively, with no significant differences between BA.2.X and BA.5.X (Fig. 3 b). Although the results obtained partially diverge from the data of earlier studies [49–52], they correspond to the dynamics of the epidemic process in Moscow. Thus, with the emergence of the Omicron variant BA.1.X, an increase in the incidence was observed, whereas BA.2.X displaced BA.1.X without an accompanying increase in the number of new COVID-19 cases (Fig. 1 a). The active spread of BA.5.X coincided with the next (6th) rise in incidence, and, theoretically, this was possible not due to an increase in viral load, but due to the appearance of a number of nonsynonymous substitutions in this variant, in particular, two additional mutations in RBD L452R and F486V, the first amino acid substitution being also characteristic of the Delta variant (Fig. 2). It has been shown that due to these mutations there is an evasion of neutralizing antibodies formed after previous infection with the first Omicron variants and vaccination [53, 54]. Furthermore, BA.5.X compared to BA.1/2.X appears to have nonsynonymous substitutions in the ORF9b (D16G) and NSP13 (T127N) genes, which may promote virus spread through evasion from the host immune system [55]. Accordingly, from the Delta variant to BA.1.X and BA.5.X, the accumulation of mutations affecting the ability to evade immunity and penetrate the cell more efficiently was consistent, which allowed these variants to cause several waves of morbidity without a corresponding increase in viral load.
Toward the end of 2022, CL.X, which is a sublineage of BA.5.1.29, becomes one of the main variants in Moscow [56]. This variant was predominantly distributed in Russia and was characterized by several additional mutations compared to the parental lineage. These mutations include K444N and K150E in the S-protein, A218S in the nucleocapsid protein, F41C in ORF8, and N102S in NSP13 (Fig. S3). Along with the occurrence of the above mutations, an increase in viral load was observed, which may indicate a positive effect of these amino acid substitutions on CL.X replication. However, these mutations were not widespread in the general population of Omicron variant virus: their proportion ranged from 0.05% (for NS8_F41C) to 0.3% (for Spike_K444N) according to GISAID data as of September 7, 2023 [57]. Due to the mutation in RBD, the virus could theoretically avoid neutralizing antibodies [58], which could promote its replication; in addition, they could potentially affect ORF8 and its role in epigenetic regulation [59] or the helicase activity of NSP13 [60]. A detailed review of the genetic structure of this lineage also revealed the T883I mutation, which appeared simultaneously with the onset of CL circulation and was present in about 29% of all collected samples belonging to the CL lineage (Fig. S3 a). The presence of T883I was associated with a higher viral load compared to the variant without substitution (Fig. S1 a), and the proportion of the CL.1.2 variant characterized by this mutation began to increase with the introduction of XBB (Fig. S2). In addition, T883I was also found in XBB.1.X in about 12% of cases, and the proportion of this mutation also began to increase in parallel with the onset of XBB.1.9.X circulation (Fig. S2), but a similar effect in terms of increased viral load was not observed for XBB.1.X + T883I compared with the variant without the substitution (Fig. S1 b). Thus, the mere presence of this mutation did not contribute to an increase in viral load; however, it is possible that the combination of mutations in the S-protein of CL.X, including T883I, could have a positive effect on virus spread, but its proportion in the population was still insufficient.
For the latter dominant variants, XBB.1.X and XBB.1.9.X, a consistent increase in viral load was also observed along with the occurrence of a number of additional amino acid substitutions in both Spike and other proteins. XBB is a recombinant of BJ.1 (sub-variant BA.2.10) and BM.1.1.1.1 (sub-variant BA.2.75) with a recombination point in the S1 of S-protein around 445–460 amino acid positions [61]. Here we observe the first case among all previously dominant SARS-CoV-2 variants of virus adaptation through recombination. The acquired mutations V83A, R346T, L368I and N460K in the S-protein have been shown to result in increased hACE2 binding affinity, fusogenicity and infectivity of the XBB variant virus [62]. These findings are consistent with our results and explain the increased viral load of XBB.1.X compared to BA.5.X and CL.X. However, according to previous studies, the presence of a stop codon at the beginning of the ORF8 gene should have had a negative effect on viral replication with a corresponding decrease in viral copies [59], but our results show the opposite effect. Perhaps the presence of several other substitutions in non-structural genes (Fig. 3) compensates for the absence of full-length NS8. The increased viral load of the XBB.1.9.X variant compared to the previously circulating XBB.1.X (Fig. 1, Fig. 3 c) may have been due to the F486P substitutions in the S-protein present in about 27% of cases, NSP3_G1001S, NSP9_T35I, NS9b_I5T. The presence of F486P was predominantly characteristic of the XBB.1.5 variant, which spread rapidly in several countries due to increased transmissibility [63] and, in agreement with earlier studies, the presence of this substitution correlated with increased affinity to hACE2 [64].
Conclusion
Molecular genetic monitoring of SARS-CoV-2 variants in Moscow since the beginning of the pandemic has allowed not only to characterize the profile of the main genetic lineages of the virus, but also to reveal several regularities that could account for the continued spread of new variants of the virus. Thus, the change of genetic variants of SARS-CoV-2 in Moscow for more than 3 years from the beginning of the pandemic was accompanied by a gradual increase in the number of mutations, where their sharp increase was characteristic of the Omicron variant that emerged at the end of 2021. Furthermore, for several successive variants, for example, from Wuhan to Delta or from BA.5 .X to XBB.1.9.X, there was a consistent increase in viral load along with the appearance of new amino acid substitutions in both the Spike protein and nonstructural proteins. In the remaining cases where this trend was not observed, there was an accumulation of several mutations that promoted evasion of previously established immunity and increased infectivity of the virus, which determined their further spread. These results can be used for modeling and predicting future variants, as they clearly demonstrate the transition: «increase in mutations ‒ increase in viral load ‒ emergence of a new serotype».
Funding. The study was funded by the Ministry of Health of the Russian Federation (Project No. 123031400022-0, titled Investigation of SARS-CoV-2 variability in relation to the biological risks associated with the reduced efficacy of therapy and prevention used during the COVID-19 pandemic).
Acknowledgement. We are grateful to all GISAID submitted and originating laboratories for access to SARS-CoV-2 sequencing data. We are grateful to Anastasia A. Zakharova and Timofey A. Remizov for their technical assistance in the implementation of the project.
Conflict of interest. The authors declare no apparent or potential conflicts of interest related to the publication of this article.
Ethics approval. The study was conducted with the informed consent of the patients. The research protocol was approved by the Ethics Committee of the National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya (Protocol No. 14 dated 29 September 2021).
About the authors
Daria D. Kustova
National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation; Federal State Budgetary Educational Institution of Higher Education Lomonosov Moscow State University
Email: kustovadaria@yandex.ru
ORCID iD: 0000-0002-8382-275X
junior researcher, Laboratory of mechanisms of population variability of pathogenic microorganisms; PhD student, Department of Virology, Faculty of Biology
Russian Federation, 123098, Moscow; 119991, MoscowAndrei A. Pochtovyi
National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation; Federal State Budgetary Educational Institution of Higher Education Lomonosov Moscow State University; I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University)
Author for correspondence.
Email: a.pochtovyy@gamaleya.org
ORCID iD: 0000-0003-1107-9351
Cand. Sci. (Biol.), senior researcher, Laboratory of mechanisms of population variability of pathogenic microorganisms
Russian Federation, 123098, Moscow; 119991, Moscow; 119435, MoscowOlga G. Shpakova
Moscow Healthcare Department
Email: shpakovaog@dcli.ru
Head of the laboratory of the Moscow Scientific and Practical Center for Laboratory Research
Russian Federation, 127006, MoscowIrina A. Shtinova
Moscow Healthcare Department
Email: shtinovaia@dcli.ru
Head of Laboratory Center of the Moscow Scientific and Practical Center for Laboratory Research
Russian Federation, 127006, MoscowNadezhda A. Kuznetsova
National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation
Email: nadyakuznetsova0@yandex.ru
ORCID iD: 0000-0002-7399-7628
Cand. Sci. (Biol.), senior researcher, Laboratory of mechanisms of population variability of pathogenic microorganisms
Russian Federation, 123098, MoscowDenis A. Kleimenov
National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation
Email: mne10000let@yandex.ru
ORCID iD: 0000-0001-9422-7238
Cand. Sci. (Med.), Head, Laboratory of translational biomedicine
Russian Federation, 123098, MoscowAndrey G. Komarov
Moscow Healthcare Department
Email: komarovag@zdrav.mos.ru
ORCID iD: 0009-0000-8597-7125
Cand. Sci. (Med.), Head of the Moscow Scientific and Practical Center for Laboratory Research
Russian Federation, 127006, MoscowVladimir A. Gushchin
National Research Centre for Epidemiology and Microbiology Named after Honorary Academician N.F. Gamaleya of the Ministry of Health of the Russian Federation; Federal State Budgetary Educational Institution of Higher Education Lomonosov Moscow State University; I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University)
Email: wowaniada@yandex.ru
ORCID iD: 0000-0002-9397-3762
Dr. Sci. (Biology), Head, Laboratory of mechanisms of population variability of pathogenic microorganisms, Reference center for coronavirus infection; senior researcher, Department of virology, Biological faculty
Russian Federation, 123098, Moscow; 119991, Moscow; 119435, MoscowReferences
- COVID-19 epidemiological update – 12 April 2024. Available at: https://who.int/publications/m/item/covid-19-epidemiological-update-edition-166
- CDC: SARS-CoV-2 Variant Classifications and Definitions. Available at: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html
- Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020; 383(27): 2603–15. https://doi.org/10.1056/NEJMoa2034577
- Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021; 384(5): 403–16. https://doi.org/10.1056/nejmoa2035389
- Logunov D.Y., Dolzhikova I.V., Shcheblyakov D.V., Tukhvatulin A.I., Zubkova O.V., Dzharullaeva A.S., et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet. 2021; 397(10275): 671–81. https://doi.org/10.1016/S0140-6736(21)00234-8
- Voysey M., Clemens S.A.C., Madhi S.A., Weckx L.Y., Folegatti P.M., Aley P.K., et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021; 397(10269): 99–111. https://doi.org/10.1016/S0140-6736(20)32661-1
- Lau J.J., Cheng S.M.S., Leung K., Lee C.K., Hachim A., Tsang L.C.H., et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA.2 variant in a SARS-CoV-2 infection-naive population. Nat. Med. 2023; 29(2): 348–57. https://doi.org/10.1038/s41591-023-02219-5
- Kurhade C., Zou J., Xia H., Liu M., Chang H.C., Ren P., et al. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023; 29(2): 344–7. https://doi.org/10.1038/s41591-022-02162-x
- Miteva D., Kitanova M., Batselova H., Lazova S., Chervenkov L., Peshevska-Sekulovska M., et al. The end or a new era of development of SARS-CoV-2 virus: genetic variants responsible for severe COVID-19 and clinical efficacy of the most commonly used vaccines in clinical practice. Vaccines (Basel). 2023; 11(7): 1181. https://doi.org/10.3390/vaccines11071181
- Munro A.P.S., Janani L., Cornelius V., Aley P.K., Babbage G., Baxter D., et al. Safety and immunogenicity of seven COVID-19 vaccines as a third dose (booster) following two doses of ChAdOx1 nCov-19 or BNT162b2 in the UK (COV-BOOST): a blinded, multicentre, randomised, controlled, phase 2 trial. Lancet. 2021; 398(10318): 2258–76. https://doi.org/10.1016/S0140-6736(21)02717-3
- Chenchula S., Karunakaran P., Sharma S., Chavan M. Current evidence on efficacy of COVID-19 booster dose vaccination against the Omicron variant: A systematic review. J. Med. Virol. 2022; 94(7): 2969–76. https://doi.org/10.1002/jmv.27697
- Chalkias S., Harper C., Vrbicky K., Walsh S.R., Essink B., Brosz A., et al. A bivalent Omicron-containing booster vaccine against COVID-19. N. Engl. J. Med. 2022; 387(14): 1279–91. https://doi.org/10.1056/NEJMoa2208343
- Scheaffer S.M., Lee D., Whitener B., Ying B., Wu K., Liang C.Y., et al. Bivalent SARS-CoV-2 mRNA vaccines increase breadth of neutralization and protect against the BA.5 Omicron variant in mice. Nat. Med. 2023; 29(1): 247–57. https://doi.org/10.1038/s41591-022-02092-8
- Winokur P., Gayed J., Fitz-Patrick D., Thomas S.J., Diya O., Lockhart S., et al. Bivalent Omicron BA.1-adapted BNT162b2 booster in adults older than 55 years. N. Engl. J. Med. 2023; 388(3): 214–27. https://doi.org/10.1056/NEJMoa2213082
- Kirsebom F.C.M., Andrews N., Stowe J., Ramsay M., Lopez Bernal J. Duration of protection of ancestral-strain monovalent vaccines and effectiveness of bivalent BA.1 boosters against COVID-19 hospitalisation in England: a test-negative case-control study. Lancet Infect. Dis. 2023; 23(11): 1235–43. https://doi.org/10.1016/S1473-3099(23)00365-1
- Iketani S., Mohri H., Culbertson B., Hong S.J., Duan Y., Luck M.I., et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023; 613(7944): 558–64. https://doi.org/10.1038/s41586-022-05514-2
- Stevens L.J., Pruijssers A.J., Lee H.W., Gordon C.J., Tchesnokov E.P., Gribble J., et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022; 14(656): eabo0718. https://doi.org/10.1126/scitranslmed.abo0718
- Imai M., Ito M., Kiso M., Yamayoshi S., Uraki R., Fukushi S., et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med. 2023; 388(1): 89–91. https://doi.org/10.1056/NEJMc2214302
- Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022; 602(7898): 657–63. https://doi.org/10.1038/s41586-021-04385-3
- Wang Q., Guo Y., Iketani S., Nair M.S., Li Z., Mohri H., et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature. 2022; 608(7923): 603–8. https://doi.org/10.1038/s41586-022-05053-w
- Pochtovyi A.A., Kustova D.D., Siniavin A.E., Dolzhikova I.V., Shidlovskaya E.V., Shpakova O.G., et al. In vitro efficacy of antivirals and monoclonal antibodies against SARS-CoV-2 Omicron lineages XBB.1.9.1, XBB.1.9.3, XBB.1.5, XBB.1.16, XBB.2.4, BQ.1.1.45, CH.1.1, and CL.1. Vaccines (Basel). 2023; 11(10): 1533. https://doi.org/10.3390/vaccines11101533
- Puhach O., Meyer B., Eckerle I. SARS-CoV-2 viral load and shedding kinetics. Nat. Rev. Microbiol. 2023; 21(3): 147–61. https://doi.org/10.1038/s41579-022-00822-w
- Gushchin V.A., Pochtovyi A.A., Kustova D.D., Ogarkova D.A., Tarnovetskii I.Y., Belyaeva E.D., et al. Dynamics of SARS-CoV-2 major genetic lineages in moscow in the context of vaccine prophylaxis. Int. J. Mol. Sci. 2022; 23(23): 14670. https://doi.org/10.3390/ijms232314670
- Wickham H., François R., Henry L., Müller K., Vaughan D. dplyr: A Grammar of Data Manipulation. Available at: https://dplyr.tidyverse.org/reference/dplyr-package.html
- Wickham H. ggplot2. New York, NY: Springer; 2009. https://doi.org/10.1007/978-0-387-98141-3
- Patil I. Visualizations with statistical details: The “ggstatsplot” approach. J. Open. Source. Softw. 2021; 6(61): 3167. https://doi.org/10.21105/joss.03167
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020; 182(4): 812–27.e19. https://doi.org/10.1016/j.cell.2020.06.043
- Markov P.V., Ghafari M., Beer M., Lythgoe K., Simmonds P., Stilianakis N.I., et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023; 21(6): 361–79. https://doi.org/10.1038/s41579-023-00878-2
- Klink G.V., Safina K.R., Nabieva E., Shvyrev N., Garushyants S., Alekseeva E., et al. The rise and spread of the SARS-CoV-2 AY.122 lineage in Russia. Virus Evol. 2022; 8(1): veac017. https://doi.org/10.1093/ve/veac017
- Teyssou E., Delagrèverie H., Visseaux B., Lambert-Niclot S., Brichler S., Ferre V., et al. The Delta SARS-CoV-2 variant has a higher viral load than the Beta and the historical variants in nasopharyngeal samples from newly diagnosed COVID-19 patients. J. Infect. 2021; 83(4): e1–3. https://doi.org/10.1016/j.jinf.2021.08.027
- von Wintersdorff C.J.H., Dingemans J., van Alphen L.B., Wolffs P.F.G., van der Veer B.M.J.W., Hoebe C.J.P.A., et al. Infections with the SARS-CoV-2 Delta variant exhibit fourfold increased viral loads in the upper airways compared to Alpha or non-variants of concern. Sci. Rep. 2022; 12(1): 13922. https://doi.org/10.1038/s41598-022-18279-5
- Guo H., Jiang J., Shen S., Ge X., Fan Q., Zhou B., et al. Additional mutations based on Omicron BA.2.75 mediate its further evasion from broadly neutralizing antibodies. IScience. 2023; 26(4): 106283. https://doi.org/10.1016/j.isci.2023.106283
- Motozono C., Toyoda M., Zahradnik J., Saito A., Nasser H., Tan T.S., et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021; 29(7): 1124–36.e11. https://doi.org/10.1016/j.chom.2021.06.006
- Singh P., Sharma K., Singh P., Bhargava A., Negi S.S., Sharma P., et al. Genomic characterization unravelling the causative role of SARS-CoV-2 Delta variant of lineage B.1.617.2 in 2nd wave of COVID-19 pandemic in Chhattisgarh, India. Microb. Pathog. 2022; 164: 105404. https://doi.org/10.1016/j.micpath.2022.105404
- Sarkar P., Banerjee S., Saha S.A., Mitra P., Sarkar S. Genome surveillance of SARS-CoV-2 variants and their role in pathogenesis focusing on second wave of COVID-19 in India. Sci. Rep. 2023; 13(1): 4692. https://doi.org/10.1038/s41598-023-30815-5
- Lin X., Sha Z., Trimpert J., Kunec D., Jiang C., Xiong Y., et al. The NSP4 T492I mutation increases SARS-CoV-2 infectivity by altering non-structural protein cleavage. Cell Host Microbe. 2023; 31(7): 1170–84. https://doi.org/10.1016/j.chom.2023.06.002
- Kim S.M., Kim E.H., Anthony M., Casel B., Kim Y.I., Sun R., et al. SARS-CoV-2 variants show temperature-dependent enhanced polymerase activity in the upper respiratory tract and high transmissibility. bioRxiv. 2022. Preprint. https://doi.org/10.1101/2022.09.27.509689.
- Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., Batra N., et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Euro Surveill. 2021; 26(24): 2100509. https://doi.org/10.2807/1560-7917.ES.2021.26.24.2100509
- Earnest R., Uddin R., Matluk N., Renzette N., Turbett S.E., Siddle K.J., et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. Cell Rep. Med. 2022; 3(4): 100583. https://doi.org/10.1016/j.xcrm.2022.100583
- Fan Y., Li X., Zhang L., Wan S., Zhang L., Zhou F. SARS-CoV-2 Omicron variant: recent progress and future perspectives. Signal Transduct. Target. Ther. 2022; 7(1): 141. https://doi.org/10.1038/s41392-022-00997-x
- Sukhikh G.T., Priputnevich T.V., Ogarkova D.A., Pochtovyi A.A., Kustova D.D., Zlobin V.I., et al. Sputnik light and Sputnik V vaccination is effective at protecting medical personnel from COVID-19 during the period of Delta variant dominance. Vaccines (Basel). 2022; 10(11): 1804. https://doi.org/10.3390/vaccines10111804
- Planas D., Veyer D., Baidaliuk A., Staropoli I., Guivel-Benhassine F., Rajah M.M., et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021; 596(7871): 276–80. https://doi.org/10.1038/s41586-021-03777-9
- Simon-Loriere E., Schwartz O. Towards SARS-CoV-2 serotypes? Nat. Rev. Microbiol. 2022; 20(4): 187–8. https://doi.org/10.1038/s41579-022-00708-x
- Wu H., Xing N., Meng K., Fu B., Xue W., Dong P., et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021; 29(12): 1788–801. https://doi.org/10.1016/j.chom.2021.11.005
- Bills C.J., Xia H., Chen J.Y.C., Yeung J., Kalveram B.K., Walker D., et al. Mutations in SARS-CoV-2 variant nsp6 enhance type-I interferon antagonism. Emerg. Microbes Infect. 2023; 12(1): 2209208. https://doi.org/10.1080/22221751.2023.2209208
- Liu Y., Liu J., Plante K.S., Plante J.A., Xie X., Zhang X., et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022; 602(7896): 294–99. https://doi.org/10.1038/s41586-021-04245-0
- Escalera A., Gonzalez-Reiche A.S., Aslam S., Mena I., Laporte M., Pearl R.L., et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe. 2022; 30(3): 373–87.e7. https://doi.org/10.1016/j.chom.2022.01.006
- Starr T.N., Greaney A.J., Hannon W.W., Loes A.N., Hauser K., Dillen J.R., et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science. 2022; 377(6604): 420–4. https://doi.org/10.1126/science.abo7896
- Hirotsu Y., Maejima M., Shibusawa M., Natori Y., Nagakubo Y., Hosaka K., et al. SARS-CoV-2 Omicron sublineage BA.2 replaces BA.1.1: Genomic surveillance in Japan from September 2021 to March 2022. J. Infect. 2022; 85(2): 174–211. https://doi.org/10.1016/j.jinf.2022.04.040
- Mastrorosa I., Cozzi-Lepri A., Colavita F., Lalle E., Mazzotta V., Cimaglia C., et al. SARS-CoV-2 nasopharyngeal viral load in individuals infected with BA.2, compared to Alpha, Gamma, Delta and BA.1 variants: A single-center comparative analysis. J. Clin. Virol. 2022; 157: 105299. https://doi.org/10.1016/j.jcv.2022.105299
- Tozer K., Sjaarda C.P., Moslinger E., Wong H., Mubareka S., Maguire F., et al. Comparison of SARS-CoV-2 viral loads in the nasal mucosa of patients infected with BA.1, BA.2, or BA.5 Omicron lineages. Open Forum Infect. Dis. 2022; 9(12): ofac564. https://doi.org/10.1093/ofid/ofac564
- Takatsuki Y., Takahashi Y., Nakajima J., Iwasaki Y., Nagano K., Tani-Sassa C., et al. Viral load of SARS-CoV-2 Omicron BA.5 is lower than that of BA.2 despite the higher infectivity of BA.5. Immun. Inflamm. Dis. 2023; 11(2): e783. https://doi.org/10.1002/iid3.783
- Cao Y., Yisimayi A., Jian F., Song W., Xiao T., Wang L., et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022; 608(7923): 593–602. https://doi.org/10.1038/s41586-022-04980-y
- Tuekprakhon A., Nutalai R., Dijokaite-Guraliuc A., Zhou D., Ginn H.M., Selvaraj M., et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell. 2022; 185(14): 2422–33. https://doi.org/10.1016/j.cell.2022.06.005
- Rashid F., Xie Z., Suleman M., Shah A., Khan S., Luo S. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 2022; 13: 940756. https://doi.org/10.3389/fimmu.2022.940756
- Github. BA.5.1.29 sublineage with S:K150E, S:460K, Orf8:F41C. Available at: https://github.com/cov-lineages/pango-designation/issues/1187
- Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., et al. GISAID’s role in pandemic response. China CDC Wkly. 2021; 3(49): 1049–51. https://doi.org/10.46234/ccdcw2021.255
- Haslwanter D., Dieterle M.E., Wec A.Z., O’Brien C.M., Sakharkar M., Florez C., et al. A combination of receptor-binding domain and N-terminal domain neutralizing antibodies limits the generation of SARS-CoV-2 spike neutralization-escape mutants. mBio. 2021; 12(5): e0247321. https://doi.org/10.1128/mBio.02473-21
- Kee J., Thudium S., Renner D.M., Glastad K., Palozola K., Zhang Z., et al. SARS-CoV-2 disrupts host epigenetic regulation via histone mimicry. Nature. 2022; 610(7931): 381–8. https://doi.org/10.1038/s41586-022-05282-z
- Hossain A., Akter S., Rashid A.A., Khair S., Alam A.S.M.R.U. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathog. 2022; 170: 105699. https://doi.org/10.1016/j.micpath.2022.105699
- Github. BJ.1/BM.1.1.1 (=BA.2.75.3.1.1.1) recombinant with breakpoint in S1 (>=5 sequences, 3x Singapore, 2x US as of 2022-09-12). Available at: https://github.com/cov-lineages/pango-designation/issues/1058
- Tamura T., Ito J., Uriu K., Zahradnik J., Kida I., Anraku Y., et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023; 14(1): 2800. https://doi.org/10.1038/s41467-023-38435-3
- Ao D., He X., Hong W., Wei X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm. (2020). 2023; 4(2): e239. https://doi.org/10.1002/mco2.239
- Parums D.V. Editorial: The XBB.1.5 (‘Kraken’) subvariant of Omicron SARS-CoV-2 and its rapid global spread. Med. Sci. Monitor. 2023; 29: e939580. https://doi.org/10.12659/MSM.939580
Supplementary files

