



Молекулярно-биологические закономерности сохранения циркуляции вируса SARS-CoV-2 в человеческой популяции
- Авторы: Кустова Д.Д.1,2, Почтовый А.А.1,2,3, Шпакова О.Г.4, Штинова И.А.4, Кузнецова Н.А.1, Клейменов Д.А.1, Комаров А.Г.4, Гущин В.А.1,2,3
-
Учреждения:
- ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России
- ФГБОУ ВО «Московский государственный университет имени М.В. Ломоносова»
- ФГАОУ ВО «Первый Московский государственный медицинский университет имени И.М. Сеченова» Минздрава России (Сеченовский Университет)
- Департамент здравоохранения Москвы
- Выпуск: Том 69, № 4 (2024)
- Страницы: 329-340
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://virusjour.crie.ru/jour/article/view/16651
- DOI: https://doi.org/10.36233/0507-4088-242
- EDN: https://elibrary.ru/uxnluj
- ID: 16651

Цитировать

Аннотация
Введение. Вирус SARS-CoV-2, являющийся возбудителем COVID-19, циркулирует в человеческой популяции 4 года. При этом уже к концу 2-го года не осталось иммунологически наивных лиц в результате активной иммунизации населения вакцинами и естественного контакта с вирусом. Понимание закономерностей, способствующих сохранению вируса в человеческой популяции, позволит лучше прогнозировать эпидемический потенциал COVID-19 и своевременно адаптировать средства противодействия продолжающейся пандемии.
Целью данной работы являлось описание выявленных молекулярно-биологических закономерностей, способствующих сохранению вируса в человеческой популяции.
Материалы и методы. В течение более 3 лет с начала пандемии COVID-19 проводили молекулярно-генетический мониторинг SARS-CoV-2, включавший в себя сбор назофарингеальных мазков от инфицированных, оценку вирусной нагрузки и последующее полногеномное секвенирование.
Результаты. Был установлен профиль доминирующих генетических линий на фоне роста заболеваемости и его изменение на протяжении всего периода мониторинга. Дополнительно изучен состав аминокислотных замен в различных белках SARS-CoV-2 и уровень вирусной нагрузки в составе мазков у последовательно сменявшихся вариантов возбудителя COVID-19. Предложена модель сохранения вируса в человеческой популяции, в рамках которой вирус способен: 1) к периодической смене серотипа (падение вируснейтрализующей активности сывороток более чем в 10 раз); 2) к накоплению дополнительных точечных аминокислотных замен в составе RBD в пределах серотипа для частичного ухода от нейтрализующих антител (снижение вируснейтрализующей активности в 2‒3 раза) и повышения сродства к рецептору; 3) к постепенному увеличению количества выделяемого вируса на слизистых оболочках в пределах одного серотипа.
Заключение. Предложенная модель в существенной степени объясняет динамику заболеваемости COVID-19 в Москве. Для более полной характеристики наблюдаемой динамики необходимо получение популяционных данных о динамике напряженности иммунитета и нейтрализующей способности антител в отношении актуального состава генетических линий.
Ключевые слова
Полный текст
Введение
Пандемия COVID-19 была вызвана обнаруженным в конце 2019 г. новым вирусом SARS-CoV-2 и стала причиной более 774 млн случаев заболевания и более 7 млн смертей по всему миру [1]. Наблюдаемая эволюция SARS-CoV-2 привела к независимому появлению ряда генетических линий, выделяемых Всемирной организацией здравоохранения в определенные периоды как варианты, вызывающие озабоченность (variants of concern, VOC), – Альфа, Бета, Гамма, Эпсилон, Дельта и Омикрон [2], которые были связаны с подъемами заболеваемости и смертности в большинстве стран. Несмотря на оперативную разработку и внедрение эффективных профилактических препаратов, по итогам первого года пандемии [3–6], в связи с появлением первых VOC, стали появляться сообщения о снижении эффективности вакцинации [7–9]. Это стало основанием для введения бустерных доз [10, 11]. В случае варианта Омикрон эффективность снизилась настолько, что пришлось вводить бустерные дозы с измененным составом [12–15]. Однако в случае относительно недавно возникшего варианта XBB даже с применением обновленного двухвалентного бустера, а затем моновалетной вакцины, содержащей антиген XBB, наблюдается снижение нейтрализации [8]. Кроме того, на данный момент накоплен большой объем информации о влиянии характерных для доминирующих вариантов мутаций на эффективность терапевтических препаратов, включающих в себя моноклональные антитела и противовирусные средства [16–18], что привело к потере эффективности большинства моноклональных антител (monoclonal antibodies, mAbs) в отношении актуально циркулирующих вариантов [19–21]. Появлению нового варианта вируса предшествовало возникновение мутаций, что в свою очередь сказывалось на изменении трансмиссивности и/или патогенности вируса, чувствительности к терапевтическим препаратам и способности уклоняться от естественного или вакциноиндуцированного иммунитета. Стоит отметить, что такие показатели, как трансмиссивность и/или патогенность вируса, неразрывно связаны с величиной вирусной нагрузки. Как обсуждалось ранее [22], хозяин (роль вакцинации или предшествующей инфекции) и вирусные факторы (варианты SARS-CoV-2) значимо влияют на динамику вирусной нагрузки и, следовательно, дополнительно влияют на передачу вируса. Несмотря на накопленные данные о значениях некоторых мутаций и связанных с ними фенотипических признаках [22], остаются не до конца изучены факторы, оказывающие влияние на динамику эпидемиологического процесса в разные периоды.
В основе настоящего исследования лежат результаты проводимого с конца 2020 г. молекулярно-генетического мониторинга вариантов вируса SARS-CoV-2, часть из которых была ранее опубликована [23]. В данной работе была проведена оценка как динамики циркулирующих вариантов, так и потенциального влияния мутаций и вирусной нагрузки как основных факторов, способствующих изменению профиля циркулирующих линий, что может отражаться в наблюдаемом переходе «увеличение мутаций ‒> увеличение вирусной нагрузки ‒> появление нового серотипа».
Материалы и методы
В период появления варианта Дельта назофарингеальные мазки собирали регулярно у разных групп добровольцев, включая тех, кто был первично инфицирован или вакцинирован. От всех пациентов было получено письменное согласие в соответствии с приказом Минздрава России от 21 июля 2015 г. № 474н «О порядке дачи информированного добровольного согласия на оказание медицинской помощи в рамках клинической апробации методов профилактики, диагностики, лечения и реабилитации, формах информированного добровольного согласия на оказание медицинской помощи в рамках клинической апробации методов профилактики, диагностики, лечения и реабилитации и отказа от медицинской помощи в рамках клинической апробации методов профилактики, диагностики, лечения и реабилитации». Все образцы были деперсонифицированы до их получения исследовательской группой. Исследование одобрено локальным этическим комитетом НИЦЭМ им. Н.Ф. Гамалеи (протокол № 14, 29 сентября 2021 г.).
Сбор образцов и оценку вирусной нагрузки выполняли в двух лабораториях (далее Лаборатория 1 и Лаборатория 2). В период циркуляции вариантов Ухань, Дельта и Омикрон (BA.1.X and BA.2.X) (Лаборатория 1) мазки из носоглотки собирали в среду для транспортировки вирусов (G00155, ГЕМ, Россия). Тотальную РНК выделяли с помощью набора QIAamp Viral RNA Mini Kit (Qiagen, Германия) и набора для выделения тотальной РНК из клеток животных и бактерий, мазков и вирусов на колонках (RU-250, «Биолабмикс», Новосибирск, Россия). Количественную полимеразную цепную реакцию (ПЦР) с обратной транскрипцией проводили с использованием набора для RT-PCR SARS-CoV-2 FRT (EA-128, НИЦЭМ им. Н.Ф. Гамалеи, Москва, Россия). Образцы со значениями Ct < 30 были отобраны для полногеномного секвенирования.
В Лаборатории 2 мазки из носоглотки собирали в транспортную среду для вирусов (физиологический раствор или транспортная среда XK-PCR30 (Jiangsu Xinkang Medical Instrumet Co., Ltd., Китай)) или транспортную среду ГЕМ (ГЕМ, Москва, Россия). В период циркуляции вариантов Омикрон BA.1.X, BA.2.X, BA.5.X количественную ПЦР с обратной транскрипцией проводили с использованием набора AmpliPrime SARS-CoV-2 DUO (NextBio, Москва, Россия) в соответствии с инструкциями производителя. В период циркуляции вариантов Омикрон BA.5.X, CL.X, XBB.1.X и XBB.1.9.X количественную ПЦР с обратной транскрипцией осуществляли с использованием CoV-2 test (TestGen, Ульяновск, Россия) в соответствии с инструкцией производителя.
Дальнейшее полногеномное секвенирование выполняли с использованием технологий Ion Torrent (Thermo Fisher Scientific, США), Illumina (Illumina, США) и Oxford Nanopore (Oxford, Великобритания). Процедуры подготовки и анализа образцов были подробно описаны ранее [21, 23]. Полученные в ходе настоящего исследования полногеномные последовательности были загружены в базу данных GISAID под следующими номерами: EPI_ISL_1710849-1710866, EPI_ISL_2296111-2296286, EPI_ISL_2296288-2296379, EPI_ISL_4572812, EPI_ISL_5334362-5334371, EPI_ISL_5334374-5334389, EPI_ISL_7211325-7211326, EPI_ISL_7263932-7263933, EPI_ISL_9230058-9230062, EPI_ISL_9230064-9230100, EPI_ISL_10627062, EPI_ISL_11864996-11865125, EPI_ISL_11872910, EPI_ISL_421275, EPI_ISL_454732, EPI_ISL_470896-470904, EPI_ISL_572398, EPI_ISL_872628-872643, EPI_ISL_875515, EPI_ISL_1015362, EPI_ISL_1708507-1708509, EPI_ISL_12225322, EPI_ISL_12748381-12748382, EPI_ISL_13431664-13431687, EPI_ISL_14217225-14217226, EPI_ISL_15327072-15327075, EPI_ISL_15858138-15859137, EPI_ISL_15860713-15860737, EPI_ISL_15860739-15860839, EPI_ISL_15860841-15860991, EPI_ISL_15860993-15861048, EPI_ISL_15862338-15863336, EPI_ISL_15863677-15864655, EPI_ISL_15864802-15865776, EPI_ISL_15865821-15866801, EPI_ISL_15867150-15868141, EPI_ISL_15868158-15869145, EPI_ISL_15869218-15870209, EPI_ISL_15871156-15872140, EPI_ISL_15872157-15873150, EPI_ISL_15873159-15874146, EPI_ISL_15874159-15875146, EPI_ISL_15875638-15876623, EPI_ISL_15876640-15877626, EPI_ISL_15879747-15880730, EPI_ISL_15883551-15884536, EPI_ISL_15884833-15885823, EPI_ISL_15885995-15886980. Также полученные последовательности были загружены в базу данных VGARus.
Информация о циркуляции генетических линий, использованная в настоящем исследовании, была дополнена данными, полученными из базы данных GISAID. Полученные данные были отфильтрованы по следующим параметрам: Host == «Human», `Sequence length` >= 27000, `Is complete?` == “TRUE” и Location == “Europe/Russia/Moscow/Moscow” или “Europe/Russia/Moscow” (дата запроса 18 сентября 2023 г.). Дополнительно полученные последовательности также были отфильтрованы по дате сбора (`Collection date` < «2023-06-01»), в результате чего была получена 16 541 запись.
Информация о генетических вариантах для оставшихся последовательностей была сгруппирована следующим образом: Ухань/Wuhan (B.1 + B.1.X), Альфа/Alpha (B.1.1.7 + Q.X), Бета/Beta (B.1.351), Дельта/Delta (B.1.617.X + AY.X), BA.1.X (B.1.1.529 + BA.1.X), BA.2.X, BA.5.X (BA.5.X + BF.X + BE.X), CL.X, XBB.1.X (без XBB.1.9.X и XBB.1.16.X), XBB.1.9.X, XBB.1.16.X (XBB.1.16.X + FU.X) и «Другие XBB»/«Other XBB» (Другие линии XBB + EG.X). Кроме того, были добавлены генетические линии B.1.1.317 и B.1.1.523. Остальные линии были объединены в группу «Другие»/«Other». Полученные данные были объединены с метаданными и визуализированы в среде R при помощи пакетов dplyr v. 1.1.1 [24] и ggplot2 v. 3.4.2 [25].
Для сравнения более двух независимых групп был использован тест Краскела–Уоллиса. Если результаты были статистически значимыми, применяли тест Данна для определения различия групп. Сравнение значений порогового цикла ПЦР проводили среди образцов, где был использован одинаковый набор для количественной ПЦР с обратной транскрипцией. Для статистического анализа был использован пакет ggstatsplot v. 0.12.1 [26] и R v. 4.2.2.
Результаты
Динамика генетических вариантов SARS-CoV-2 в Москве с начала пандемии COVID-19
С начала распространения SARS-CoV-2 в Москве и до февраля 2021 г. основным доминирующим вариантом являлся Ухань (B.1.X), причем среди множества входящих в него генетических линий была отмечена циркуляция характерной преимущественно для России генетической линии B.1.1.317. Зимой 2021 г. ранее циркулировавшие варианты начали постепенно замещаться линиями Альфа (B.1.1.7 + Q*) и B.1.1.523 (также характерной в основном для России), которые в мае 2021 г. вместе с очередным подъемом заболеваемости COVID-19 были вытеснены вариантом Дельта (B.1.617.2 + AY*). В конце 2021 г. на смену варианту Дельта приходит Омикрон (B.1.1.529), и далее наблюдалась последовательная смена генетических линий BA.1.X, BA.2.X и BA.5.X. В октябре 2022 г. одним из доминирующих вариантов становится CL.X, столь широкое распространение которого не было зарегистрировано в других странах. Наряду с CL.X в ноябре 2022 г. в Москве начал циркулировать рекомбинантный вариант XBB, впоследствии вытеснивший остальные генетические линии SARS-CoV-2 (рис. 1 а).

Рис. 1. Генетические варианты SARS-CoV-2, циркулировавшие в Москве с начала пандемии COVID-19. a – динамика генетических вариантов SARS-CoV-2. Слева по оси ординат показана доля генетических линий, справа – число новых случаев COVID-19 на 100 тыс. населения; б – изменение количества мутаций в основных циркулировавших вариантах SARS-CoV-2. По оси ординат показано количество мутаций. На оси абсцисс отражены периоды (месяцы).
Fig. 1. Genetic variants of SARS-CoV-2 circulating in Moscow since the start of the COVID-19 pandemic. a – dynamics of genetic variants of SARS-CoV-2 circulating in Moscow. The left Y-axis indicates the proportion of genetic lineages, while the right axis represents the number of new COVID-19 cases per 100,000 population; b – changes in the number of mutations in the predominant circulating variants of SARS-CoV-2. The ordinate axis displays the mutation count. The periods (months) are indicated on the abscissa axis.
Разные варианты вируса, получавшие относительно широкое распространение, обладали характерным профилем мутаций. Рассмотрение общего их количества в S-белке и других генах показало, что до появления варианта Омикрон происходило постепенное их увеличение; распространение последнего же характеризовалось более чем двукратным увеличением числа несинонимичных мутаций в геноме SARS-CoV-2 (рис. 1 б). Далее в вирусе также продолжили появляться замены, однако из общего ряда «выпадает» первый вариант Омикрона, BA.1.X, характеризующийся 34 мутациями в S-белке, что на 3 больше, чем у последующего BA.2.X, и сопоставимо с BA.5.X. Несмотря на этот факт, общее число мутаций в геноме SARS-CoV-2 варианта BA.1.X было наименьшим среди всех остальных линий Омикрона и составило 60, тогда как последний рассматриваемый вариант, XBB.1.9.X, имел около 80 аминокислотных замен, из которых 41 находилась в S-белке.
Профиль мутаций в основных генетических вариантах SARS-CoV-2.
Подробное рассмотрение мутаций в выбранных вариантах выявило характерный профиль мутаций для циркулировавших в Москве вариантов SARS-CoV-2. В RBD S-белка для Альфа отмечалась мутация N501Y, для B.1.1.523 – E484K и S494P. Для Дельта были характерны две аминокислотные замены (L452R и T478K) в RBD S-белка, для BA.1.X – 13, причем также наблюдалось появление в относящихся к данному варианту Омикрона некоторых генетических линиях мутаций K417N, N440K и G446S. Для варианта BA.2.X было характерно 16 мутаций в RBD, где общими с BA.1.X являлись лишь 10 из них: далее у доминирующих вариантов не отмечались мутации R346K и S371L, присутствовавшие у BA.1.X, и, кроме того, появился ряд дополнительных замен S371F, T376A, D405N, R408S, K417N и N440K. У вариантов BA.5.X и CL.X появилась мутация L452R, характерная для линии Дельта, и F486V, причем для всех в дальнейшем циркулировавших вариантов были также характерны аминокислотные замены в положении 486. С конца 2022 г. в Москве доминирующей генетической линией стала XBB и ее подварианты (рис. 1) с характерными минимум 22 аминокислотными мутациями в RBD, причем из них 6 не встречались в ранее циркулировавших вариантах (рис. 2). Весной 2023 г. основным циркулирующим вариантом являлся XBB.1.9.X, а именно XBB.1.9.1, для которого характерна замена F486P (рис. 2).

Рис. 2. Мутации в RBD Spike-белка основных доминировавших вариантов SARS-CoV-2. По оси ординат отражены варианты SARS-CoV-2, по оси абсцисс – аминокислотные замены. Нулевые значения отражают долю мутаций, стремящуюся к нулю; пустые ячейки – отсутствие мутаций в данной позиции.
Fig. 2. Mutations in the RBD of the spike protein of the predominant SARS-CoV-2 variants. The Y-axis represents SARS-CoV-2 variants, and the X-axis represents amino acid substitutions. Zero values reflect the proportion of mutations tending to zero; empty cells represent the absence of mutations at a specific position.
В N-концевом домене (NTD) S-белка наблюдалось большое разнообразие аминокислотных замен. Генетическая линия B.1.1.523 характеризовалась наличием мутации F306L. Генетические линии, относящиеся к варианту Дельта, имели мутации, которые впоследствии не встречались у других вариантов: T19R, E156G, F157del, R158del, и одну мутацию G142D, характерную для всех вариантов линии Омикрон, кроме BA.1.X. В целом большинство мутаций BA.1.X были нехарактерны для последующих вариантов Омикрона за исключением делеций 69‒70, которые присутствовали также у BA.5.X и CL.X. CL.X, по сравнению с BA.5.X и другими вариантами Омикрона, обладал аминокислотной заменой K150E, в остальном же варианты BA.2.X, BA.5.X и CL.X имели сходный профиль мутаций. Для вариантов XBB были характерны мутации V83A, H146Q, Q183E и V213E, ранее не встречавшиеся у других генетических линий. В C-концевом домене (CTD) у всех вариантов имела место замена D614G, как известно, закрепившаяся еще до разделения генетических линий SARS-CoV-2 на VOC [27]. Для Альфа наибольшее количество аминокислотных мутаций наблюдалось именно в области CTD: P681H, T716I, S982A и D1118H, не считая D614G. Для B.1.1.523 было характерно наличие D839V и T1027I, далее не отмечавшееся в других вариантах. У Дельта в CTD было лишь 2 замены помимо D614G: P681R около фуринового сайта и D950N. Далее BA.1.X обладал наибольшим количеством мутаций в этой области по сравнению с остальными вариантами Омикрона: помимо характерных H655Y, N679K, N681H, N764K, D796Y, Q954H, N969K у первого варианта Омикрона были дополнительные T547K, N856K и L981F. В целом дальнейшая эволюция NTD Омикрон практически не была отмечена, однако у CL.X и XBB.1.X в небольшом проценте случаев (29 и 12 соответственно) наблюдалась замена T883I (рис. S3 а).
В неструктурных генах частота возникновения мутаций была значительно ниже, чем в S-белке. Для варианта Дельта было характерно около 16 мутаций в неструктурных генах, причем мутации T492I в NSP4 и P323L в RdRp (NSP12) были также характерны для всех последующих вариантов Омикрона, а G671S в том же гене – для генетических линий XBB. Остальные же мутации далее не встречались. Первый вариант Омикрона, BA.1.X, обладал аминокислотными заменами NSP3_K38R, NSP3_L1266I, NSP3_A1892T, NSP6_I189V и делециями NSP3_S1265del и NSP6_L105del, далее не встречавшимися у вариантов Омикрона, а также рядом других характерных для Омикрона мутаций: NS9b_P10S, NS9b_E27-29del, NSP4_T492I, NSP5_P132H, NSP6_S106-107del и NSP14_I42V. Все последующие варианты Омикрона по сравнению с BA.1.X обладали мутациями NS3a_T223I, NSP1_S135R, NSP3_T24I, NSP3_G489S, NSP4_L264F, NSP4_T327I, NSP6_F108del, NSP13_R392C, NSP15_T112I. Отличительной особенностью BA.2.X и XBB являлись аминокислотные замены в NS6 – D61L и NSP4 – L438F. Для BA.5.X характерны дополнительные мутации NS9b_D16G и NSP13_T127N, не встречавшиеся у других вариантов. CL.X, хоть и являясь подлинией BA.5.X, не обладал данными заменами, однако характеризовался наличием NS8_F41C и NSP13_N102S. Варианты XBB, помимо перечисленных мутаций, обладали стоп-кодоном в 8-м положении NS8 и NSP1_K47R, а XBB.1.9.X – NS9b_I5T, NSP3_G1001S, NSP9_T35I и в небольшом проценте случаев – NSP2_D449E (рис. S3 в).
Что касается других структурных генов SARS-CoV-2, то для варианта Дельта не были характерны мутации в E-белке, однако наблюдались замены в матриксном белке (M_I82T) и нуклеокапсидном (N_D63G, N_R203M, N_G215C, N_D377Y), причем, как и в случае с большинством остальных аминокислотных замен, они не встречались у Омикрона. Для всех вариантов Омикрона, циркулировавших в Москве, были характерны следующие мутации: E_T9I, M_Q19E, M_A63T, N_P13L, N_E31-33del, N_R203K, N_G204R, однако некоторые генетические линии имели свои особенности. Так, BA.1.X нес дополнительную к перечисленным мутацию M_D3G, вариант BA.2.X и все остальные рассматриваемые варианты Омикрона – N_S413R, BA.5.X и CL.X – M_D3N. CL.X также обладал заменой N_A218S, а все XBB – E_T11A (рис. S3 б).
Динамика вирусной нагрузки основных циркулировавших вариантов
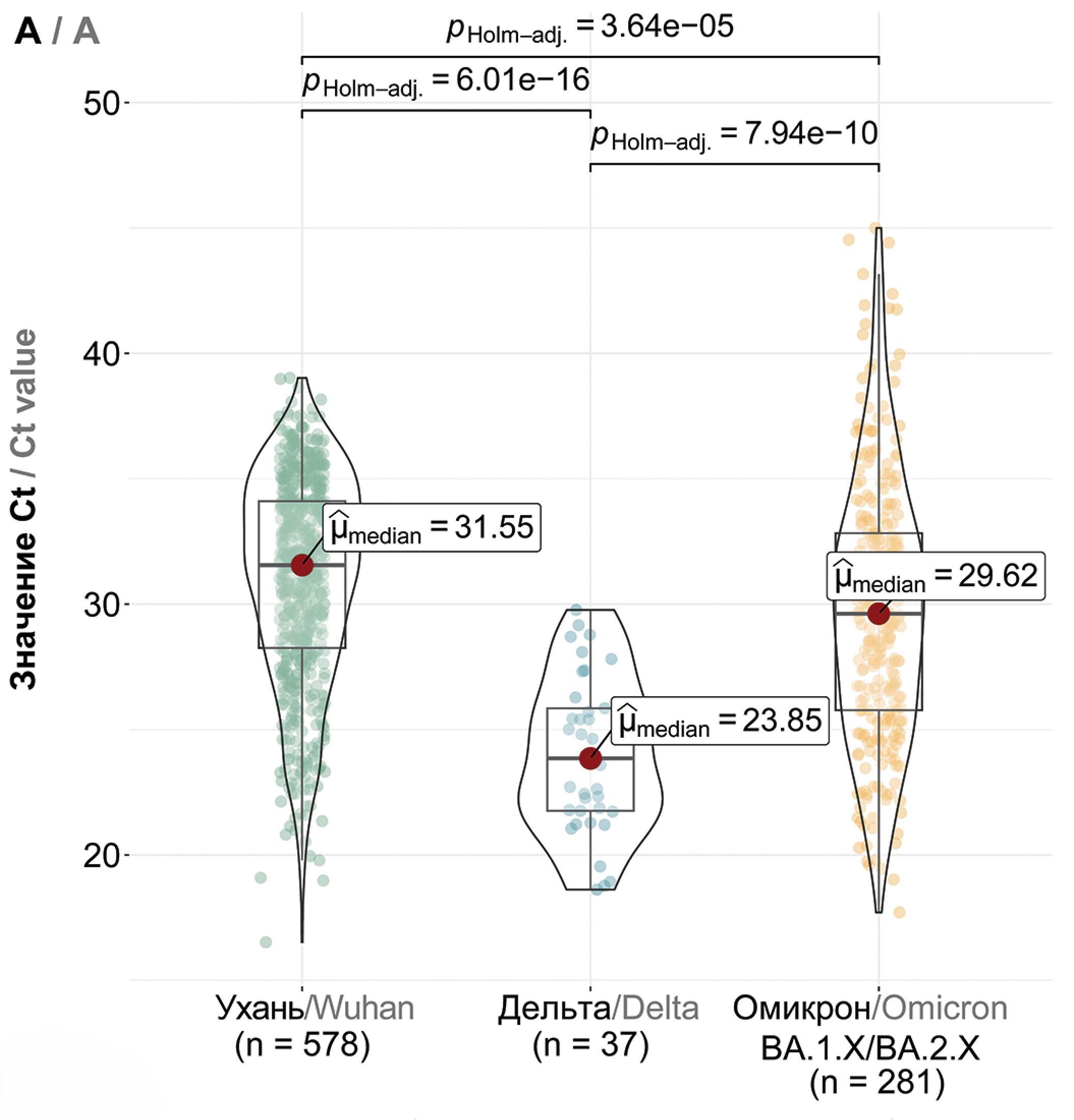
Сравнительный анализ вирусной нагрузки (эквивалент – пороговый цикл ПЦР, Ct) показал, что среди вариантов Ухань (B.1.X), Дельта (B.1.617.2 + AY.X) и первых вариантов Омикрона (BA.1/BA.2) наибольшая вирусная нагрузка была характерна для варианта Дельта с медианным значением 23,85 (p < 0,0001, рис. 3 a). Таким образом, наблюдалось увеличение вирусной нагрузки при замещении варианта Ухань генетическими линиями, относящихся к варианту Дельта (в пределах одного «серотипа»). Первые варианты Омикрона характеризовались сниженной по сравнению с Дельта вирусной нагрузкой (медиана 29,62, p<0,0001), причем далее также наблюдалось статистически достоверное небольшое повышение значений Ct для вариантов BA.2.X и BA.5.X по сравнению с BA.1.X (рис. 3 б). Статистически значимых различий между BA.2.X и BA.5.X обнаружено не было. Для варианта CL.X было характерно сниженное по сравнению с BA.5.X значение Ct с медианными значениями 35,13 и 27,27 соответственно (p<0,0001, рис. 3 в). Кроме того, у вариантов вируса CL с дополнительной мутацией T883I наблюдалась более высокая вирусная нагрузка по сравнению с вариантами без данной мутации (p=0,001, рис. S1), однако на вирусную нагрузку XBB.1.X наличие данной мутации не влияло (рис. S1). Была отмечена тенденция к увеличению вирусной нагрузки у последовательно сменяющихся вариантов BA.5.X, CL.X, XBB.1.X и XBB.1.9.X, где наибольшая вирусная нагрузка была характерна для XBB.1.9.X (p<0,01, рис. 3 в).

Рис. 3. Сравнение вирусной нагрузки основных доминировавших в Москве вариантов SARS-CoV-2. a – вирусная нагрузка вариантов Ухань (B.1.X), Дельта (B.1.617.2 + AY.X) и Омикрон (BA.1/BA.2.X); б – вирусная нагрузка вариантов Омикрон BA.1.X, BA.2.X и BA.5.X; в – вирусная нагрузка вариантов BA.5.X, CL.X, XBB.1.X и XBB.1.9.X. По оси ординат отражены значения Ct, по оси абсцисс – варианты SARS-CoV-2 и количество образцов.
Fig. 3. Comparative analysis of viral load in predominant SARS-CoV-2 variants in Moscow. a – viral load in Wuhan (B.1.X), Delta (B.1.617.2 + AY.X), and Omicron (BA.1/BA.2.X) variants; b – viral load in Omicron BA.1.X, BA.2.X, and BA.5.X variants; c – viral load in BA.5.X, CL.X, XBB.1.X, and XBB.1.9.X variants. The Y-axis denotes Ct values, while the X-axis represents SARS-CoV-2 variants and the number of samples.
Обсуждение
На протяжении более 4 лет пандемии COVID-19 был получен беспрецедентный объем данных, прежде всего геномных, которые используются как для понимания эволюции, так и для совершенствования профилактических и терапевтических препаратов. В течение данного периода появлялись варианты вируса с разными фенотипическими характеристиками, включая заразность, тяжесть заболевания и уклонение от иммунитета [28]. Комплексный анализ полученных данных о патогене SARS-CoV-2 позволяет понять основные механизмы (драйверы) эволюции и осознать процессы, порождающие это разнообразие, чтобы потенциально предсказать возможные будущие варианты вируса. В качестве основных драйверов продолжающейся пандемии нами были рассмотрены два ключевых аспекта – вирусная нагрузка и профиль мутаций в геноме вируса, что в целом характеризует наблюдаемый переход «увеличение мутаций ‒> увеличение вирусной нагрузки ‒> появление нового серотипа».
С начала пандемии в 2020 г. и до начала лета 2023 г. в Москве сменилось несколько генетических вариантов SARS-CoV-2, а именно Ухань (B.1.X), Дельта (B.1.617.2 + AY.X) и Омикрон. Внутри каждого из них в соответствующие периоды времени доминировали определенные генетические линии: в 2020 г. – B.1.1.317, в 2021 г. – B.1.1.523, Альфа (B.1.1.7 + Q.4) и AY.122 [29], в 2022 г. – линии BA.1.X, BA.2.X, BA.5.X, CL.X, в 2023 г. – рекомбинант XBB с соответствующими подлиниями XBB.1.X и XBB.1.9.X (рис 1 а). Закономерной особенностью стало последовательное увеличение числа несинонимичных мутаций в геноме циркулирующих вариантов SARS-CoV-2, около половины из которых находилось в S-белке (рис. 1 б). Последующее подробное рассмотрение молекулярно-генетических свойств циркулировавших вариантов SARS-CoV-2, включавших профиль мутаций в геноме SARS-CoV-2 и вирусную нагрузку, позволило выявить определенные закономерности, которые могли обуславливать продолжающееся распространение новых вариантов возбудителя COVID-19 и вновь возникающие подъемы заболеваемости и госпитализаций в России и мире. Так, было обнаружено, что при переходе от вариантов B.1.X к Дельта (B.1.618.2 + AY.X) произошло снижение вирусной нагрузки на 7,7 цикла ПЦР, причем это явление было ранее также отмечено и в других исследованиях [30, 31]. Для данного варианта также был характерен ряд мутаций, в том числе и в рецептор-связывающем домене Spike-белка (рис. 2, рис. S3), которые потенциально могли обуславливать его повышенную вирусную нагрузку. В ранее проведенных исследованиях было показано, что наличие замены L452R в S-белке коррелирует с уклонением от нейтрализующих антител [32], повышенными родством к рецептору ACE2, а также увеличивает стабильность шипов и вирусную инфекционность, тем самым способствуя увеличению вирусной репликации [33]. Кроме того, мутации E156G, T478K и D614G также увеличивали инфекционность и сродство вируса к рецептору, а другие замены, такие как P681R и D950N, способствовали более высокой скорости распространения за счет более эффективного расщепления S1/S2 в фуриновом сайте [34, 35]. Помимо структурных белков, на распространение варианта Дельта и повышение его вирусной нагрузки соответственно также могли оказывать влияние и другие мутации. Например, было показано, что мутация T492I в NSP4 положительно влияла на репликацию вируса [36]. Варианты вируса с аминокислотной заменой P323L в гене RdRp in vitro обладали селективным преимуществом по сравнению с вариантами без нее, и, кроме того, наличие L323 и S671 было связано с более эффективной репликацией при пониженной температуре в верхних дыхательных путях [37]. Таким образом, дополнительные мутации, характерные для варианта Дельта, непосредственно могли обусловить высокую вирусную нагрузку, в результате чего ее повышенные значения по сравнению с первыми вариантами SARS-CoV-2 B.1.X могли определять более высокую скорость распространения данного варианта в сравнении с ранее циркулировавшими [23, 38, 39].
Первые варианты Омикрона, BA.1/BA.2.X, сменившие вариант Дельта, характеризовались, наоборот, сниженной вирусной нагрузкой при более высокой скорости распространения [40]. Стоит отметить, что Омикрон филогенетически расходится с ранее циркулировавшими вариантами и обладает более 15 заменами только в RBD (рис. 2), не считая мутаций в других генах (рис. S3). Кроме того, данный вариант серологически отличается от ранее циркулировавших, о чем свидетельствует ряд исследований. Если эффективность вакцинации против Дельта оставалась высокой [41] и большинство моноклональных антител нейтрализовали вирус [42], то в случае Омикрона наблюдалось снижение эффективности вакцинации и чувствительности к терапевтическим и профилактическим препаратам [7, 8]. Таким образом, можно предположить, что Омикрон распространялся активно на первых этапах не за счет повышенной вирусной нагрузки, а за счет того, что иммунная система ранее не сталкивалась с подобным серотипом вируса [43]. Помимо этого, приобретенные в ходе эволюции варианта Омикрон мутации в неструктурных белках (к примеру, 203K/204R в N-белке и ΔSGF/ΔLSG в NSP6) и S-белке (N501Y и H655Y) также могли способствовать его широкому распространению ввиду высокой скорости репликации и более эффективной передачи вируса [44–47]. Кроме того, резкое увеличение числа мутаций в RBD может являться следствием их взаимного влияния, где появление одной замены влекло за собой появление другой, как в случае Q498R и N501Y, что также влияло на аффинность связывания ACE2 и инфекционность вируса соответственно [48]. Со второй половины 2022 г. доминирующим вариантом Омикрона в Москве становится BA.5.X, тем самым замещая ранее циркулировавший BA.2.X (рис. 1 a). Сравнительный анализ показал, что вирусная нагрузка генетических линий BA.2.X и BA.5.X статистически достоверно отличается от BA.1.X с медианными значениями Ct 25,1, 25,0 и 24,3 соответственно при отсутствии значимых различий между BA.2.X и BA.5.X (рис. 3 б). Несмотря на то что полученные результаты частично расходятся с данными ранее проведенных исследований [49–52], они соответствуют динамике эпидемического процесса в Москве. Так, с появлением варианта Омикрона, BA.1.X, наблюдалось повышение заболеваемости, тогда как BA.2.X вытеснил BA.1.X без сопутствующего увеличения числа новых случаев COVID-19 (рис. 1 a). Активное распространение BA.5.X совпало с очередным (6-м) подъемом заболеваемости, и, теоретически, это стало возможно не за счет повышения вирусной нагрузки, а благодаря появлению у данного варианта ряда несинонимичных замен, в частности, двух дополнительных мутаций в RBD L452R и F486V, причем первая аминокислотная замена была также характерна и для варианта Дельта (рис. 2). Было показано, что за счет данных мутаций наблюдается уклонение от нейтрализующих антител, сформированных после ранее перенесенной инфекции первыми вариантами Омикрона и вакцинацией [53, 54]. Кроме того, у BA.5.X по сравнению с BA.1/2.X появляются несинонимичные замены в генах ORF9b (D16G) и NSP13 (T127N), которые могут способствовать распространению вируса благодаря уклонению от иммунной системы хозяина [55]. Соответственно, от варианта Дельта к BA.1.X и BA.5.X последовательно происходило накопление влияющих на способность ускользать от иммунитета и более эффективно проникать в клетку мутаций, что позволило данным вариантам стать причиной нескольких волн заболеваемости без соответствующего повышения вирусной нагрузки.
Ближе к концу 2022 г. в Москве одним из основных вариантом становится CL.X, являющийся подлинией BA.5.1.29 [56]. Данный вариант был распространен преимущественно в России и характеризовался рядом дополнительных по сравнению с родительской линией мутаций. К числу таких мутаций относятся K444N и K150E в S-белке, A218S в белке нуклеокапсида, F41C в ORF8 и N102S в NSP13 (рис. S3). Наряду с появлением перечисленных мутаций наблюдалось повышение вирусной нагрузки, что может свидетельствовать о положительном влиянии данных аминокислотных замен на репликацию CL.X. Однако в общей популяции вируса варианта Омикрон эти мутации не были широко распространены: их доля составляла от 0,05% (для NS8_F41C) до 0,3% (для Spike_K444N) по данным GISAID на 7 сентября 2023 г. [57]. Благодаря мутации в RBD вирус теоретически мог избегать нейтрализующих антител [58], что могло способствовать его репликации; кроме того, они потенциально могли влиять на ORF8 и его роль в эпигенетической регуляции [59] или хеликазную активность NSP13 [60]. Подробное рассмотрение генетической структуры данной линии позволило также выявить мутацию T883I, появившуюся одновременно с началом циркуляции CL и присутствующую в около 29% всех собранных образцов, относящихся к линии CL (рис. S3 а). Присутствие T883I было связано с более высокой ВН по сравнению с вариантом без замены (рис. S1 a), а доля варианта CL.1.2, для которого характерна данная мутация, начала увеличиваться с интродукцией XBB (рис. S2). Кроме того, в около 12% случаев T883I встречалась и в XBB.1.X, и также доля данной мутации начала возрастать параллельно с началом циркуляции XBB.1.9.X (риc. S2), однако схожего эффекта в виде повышения вирусной нагрузки для XBB.1.X + T883I по сравнению с вариантом без замены не наблюдалось (рис. S1 б). Таким образом, само присутствие данной мутации не способствовало повышению вирусной нагрузки, однако, возможно, что комбинация мутаций в S-белке CL.X, включающая T883I, могла положительно влиять на распространение вируса, однако ее доля в популяции была все-таки недостаточна.
Для последних доминирующих вариантов, XBB.1.X и XBB.1.9.X, также наблюдалось последовательное повышение вирусной нагрузки наряду с появлением ряда дополнительных аминокислотных замен как в Spike, так и в других белках. XBB является рекомбинантом BJ.1 (подвариант BA.2.10) и BM.1.1.1 (подвариантом BA.2.75) с точкой рекомбинации в S1 S-белка в районе 445–460 аминокислотных позиций [61]. Здесь наблюдается первый случай среди всех ранее доминировавших вариантов SARS-CoV-2 приспособления вируса за счет рекомбинации. Было показано, что благодаря приобретенным мутациям V83A, R346T, L368I и N460K в S-белке отмечается повышенная аффинность связывания hACE2, фузогенность и инфекционность вируса варианта XBB [62]. Эти данные согласуются с полученными результатами и объясняют повышенную вирусную нагрузку XBB.1.X по сравнению с BA.5.X и CL.X. Однако, в соответствии с ранее проведенными исследованиями, наличие стоп-кодона в начале гена ORF8 должно было отрицательно влиять на вирусную репликацию с соответствующим уменьшением вирусных копий [59], однако по нашим результатам наблюдается противоположный эффект. Возможно, присутствие ряда других замен в неструктурных генах (рис. S3) компенсирует отсутствие полноценного NS8. Увеличенная вирусная нагрузка варианта XBB.1.9.X по сравнению с ранее циркулировавшим XBB.1.X (рис. 1, рис. 3 в), возможно, была связана с заменами F486P в S-белке, присутствующими в примерно 27% случаев, NSP3_G1001S, NSP9_T35I, NS9b_I5T. Наличие F486P было характерно преимущественно для варианта XBB.1.5, быстро распространившегося в ряде стран ввиду повышенной трансмиссивности [63] и, в соответствии с ранее проведенными исследованиями, присутствие данной замены коррелировало с повышенным сродством к hACE2 [64].
Заключение
Молекулярно-генетический мониторинг вариантов SARS-CoV-2 в Москве с начала пандемии позволил не только охарактеризовать профиль основных генетических линий вируса, но также выявить ряд закономерностей, которые могли обусловить продолжающееся распространение новых вариантов вируса. Так, смена генетических вариантов SARS-CoV-2 в Москве на протяжении более 3 лет с начала пандемии сопровождалась постепенным возрастанием числа мутаций, где резкое их увеличение было характерно для варианта Омикрон, возникшего в конце 2021 г. Кроме того, для ряда последовательно сменявшихся вариантов, к примеру, от Ухань к Дельта или от BA.5.X к XBB.1.9.X, наблюдалось закономерное повышение вирусной нагрузки наряду с появлением новых аминокислотных замен как в Spike-белке, так и в неструктурных белках. В остальных случаях, когда не отмечалась данная тенденция, происходило накопление ряда мутаций, способствующих уклонению от ранее сформированного иммунитета и повышению инфекционности вируса, что определяло его дальнейшее распространение. Эти результаты могут быть использованы для моделирования и предсказания будущих вариантов, так как наглядно демонстрируют наблюдаемый переход «увеличение мутаций ‒> увеличение вирусной нагрузки ‒> появление нового серотипа».
Финансирование. Исследование выполнено при финансовой поддержке Министерства здравоохранения РФ в рамках реализации Государственного задания № 123031400022-0 «Изучение изменчивости SARS-CoV-2 в контексте биологических рисков снижения эффективности применяемых средств терапии и профилактики в ходе пандемии COVID-19».
Благодарности. Мы благодарны всем лабораториям, предоставляющим данные GISAID, за доступ к данным секвенирования SARS-CoV-2. Мы благодарны А.А. Захаровой и Т.А. Ремизову за техническую помощь в реализации проекта.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Этическое утверждение. Исследование проводилось при добровольном информированном согласии пациентов. Протокол исследования одобрен Этическим комитетом НИЦЭМ им. Н.Ф. Гамалеи (Протокол № 14 от 29 сентября 2021 г.).
Об авторах
Дарья Дмитриевна Кустова
ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России; ФГБОУ ВО «Московский государственный университет имени М.В. Ломоносова»
Email: kustovadaria@yandex.ru
ORCID iD: 0000-0002-8382-275X
младший научный сотрудник лаборатории механизмов популяционной изменчивости патогенных микроорганизмов; аспирант кафедры вирусологии Биологического факультета
Россия, 123098, г. Москва; 119991, г. МоскваАндрей Андреевич Почтовый
ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России; ФГБОУ ВО «Московский государственный университет имени М.В. Ломоносова»; ФГАОУ ВО «Первый Московский государственный медицинский университет имени И.М. Сеченова» Минздрава России (Сеченовский Университет)
Автор, ответственный за переписку.
Email: a.pochtovyy@gamaleya.org
ORCID iD: 0000-0003-1107-9351
канд. биол. наук, старший научный сотрудник лаборатории механизмов популяционной изменчивости патогенных микроорганизмов
Россия, 123098, г. Москва; 119991, г. Москва; 119435, г. МоскваОльга Геннадьевна Шпакова
Департамент здравоохранения Москвы
Email: shpakovaog@dcli.ru
заведующая лабораторией МНПЦЛИ
Россия, 127006, г. МоскваИрина Александровна Штинова
Департамент здравоохранения Москвы
Email: shtinovaia@dcli.ru
заведующая лабораторным центром МНПЦЛИ
Россия, 127006, г. МоскваНадежда Анатольевна Кузнецова
ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России
Email: nadyakuznetsova0@yandex.ru
ORCID iD: 0000-0002-7399-7628
канд. биол. наук, старший научный сотрудник лаборатории механизмов популяционной изменчивости патогенных микроорганизмов
Россия, 123098, г. МоскваДенис Александрович Клейменов
ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России
Email: mne10000let@yandex.ru
ORCID iD: 0000-0001-9422-7238
канд. мед. наук, заведующий лабораторией трансляционной биомедицины
Россия, 123098, г. МоскваАндрей Григорьевич Комаров
Департамент здравоохранения Москвы
Email: komarovag@zdrav.mos.ru
ORCID iD: 0009-0000-8597-7125
канд. мед. наук, директор МНПЦЛИ
Россия, 127006, г. МоскваВладимир Алексеевич Гущин
ФГБУ «Национальный исследовательский центр эпидемиологии и микробиологии им. почетного академика Н.Ф. Гамалеи» Минздрава России; ФГБОУ ВО «Московский государственный университет имени М.В. Ломоносова»; ФГАОУ ВО «Первый Московский государственный медицинский университет имени И.М. Сеченова» Минздрава России (Сеченовский Университет)
Email: wowaniada@yandex.ru
ORCID iD: 0000-0002-9397-3762
д-р биол. наук, заведующий лабораторией механизмов популяционной изменчивости патогенных микроорганизмов и референс-центра по коронавирусной инфекции; старший научный сотрудник кафедры вирусологии Биологического факультета
Россия, 123098, г. Москва; 119991, г. Москва; 119435, г. МоскваСписок литературы
- COVID-19 epidemiological update – 12 April 2024. Available at: https://who.int/publications/m/item/covid-19-epidemiological-update-edition-166
- CDC: SARS-CoV-2 Variant Classifications and Definitions. Available at: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html
- Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020; 383(27): 2603–15. https://doi.org/10.1056/NEJMoa2034577
- Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021; 384(5): 403–16. https://doi.org/10.1056/nejmoa2035389
- Logunov D.Y., Dolzhikova I.V., Shcheblyakov D.V., Tukhvatulin A.I., Zubkova O.V., Dzharullaeva A.S., et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet. 2021; 397(10275): 671–81. https://doi.org/10.1016/S0140-6736(21)00234-8
- Voysey M., Clemens S.A.C., Madhi S.A., Weckx L.Y., Folegatti P.M., Aley P.K., et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021; 397(10269): 99–111. https://doi.org/10.1016/S0140-6736(20)32661-1
- Lau J.J., Cheng S.M.S., Leung K., Lee C.K., Hachim A., Tsang L.C.H., et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA.2 variant in a SARS-CoV-2 infection-naive population. Nat. Med. 2023; 29(2): 348–57. https://doi.org/10.1038/s41591-023-02219-5
- Kurhade C., Zou J., Xia H., Liu M., Chang H.C., Ren P., et al. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023; 29(2): 344–7. https://doi.org/10.1038/s41591-022-02162-x
- Miteva D., Kitanova M., Batselova H., Lazova S., Chervenkov L., Peshevska-Sekulovska M., et al. The end or a new era of development of SARS-CoV-2 virus: genetic variants responsible for severe COVID-19 and clinical efficacy of the most commonly used vaccines in clinical practice. Vaccines (Basel). 2023; 11(7): 1181. https://doi.org/10.3390/vaccines11071181
- Munro A.P.S., Janani L., Cornelius V., Aley P.K., Babbage G., Baxter D., et al. Safety and immunogenicity of seven COVID-19 vaccines as a third dose (booster) following two doses of ChAdOx1 nCov-19 or BNT162b2 in the UK (COV-BOOST): a blinded, multicentre, randomised, controlled, phase 2 trial. Lancet. 2021; 398(10318): 2258–76. https://doi.org/10.1016/S0140-6736(21)02717-3
- Chenchula S., Karunakaran P., Sharma S., Chavan M. Current evidence on efficacy of COVID-19 booster dose vaccination against the Omicron variant: A systematic review. J. Med. Virol. 2022; 94(7): 2969–76. https://doi.org/10.1002/jmv.27697
- Chalkias S., Harper C., Vrbicky K., Walsh S.R., Essink B., Brosz A., et al. A bivalent Omicron-containing booster vaccine against COVID-19. N. Engl. J. Med. 2022; 387(14): 1279–91. https://doi.org/10.1056/NEJMoa2208343
- Scheaffer S.M., Lee D., Whitener B., Ying B., Wu K., Liang C.Y., et al. Bivalent SARS-CoV-2 mRNA vaccines increase breadth of neutralization and protect against the BA.5 Omicron variant in mice. Nat. Med. 2023; 29(1): 247–57. https://doi.org/10.1038/s41591-022-02092-8
- Winokur P., Gayed J., Fitz-Patrick D., Thomas S.J., Diya O., Lockhart S., et al. Bivalent Omicron BA.1-adapted BNT162b2 booster in adults older than 55 years. N. Engl. J. Med. 2023; 388(3): 214–27. https://doi.org/10.1056/NEJMoa2213082
- Kirsebom F.C.M., Andrews N., Stowe J., Ramsay M., Lopez Bernal J. Duration of protection of ancestral-strain monovalent vaccines and effectiveness of bivalent BA.1 boosters against COVID-19 hospitalisation in England: a test-negative case-control study. Lancet Infect. Dis. 2023; 23(11): 1235–43. https://doi.org/10.1016/S1473-3099(23)00365-1
- Iketani S., Mohri H., Culbertson B., Hong S.J., Duan Y., Luck M.I., et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023; 613(7944): 558–64. https://doi.org/10.1038/s41586-022-05514-2
- Stevens L.J., Pruijssers A.J., Lee H.W., Gordon C.J., Tchesnokov E.P., Gribble J., et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022; 14(656): eabo0718. https://doi.org/10.1126/scitranslmed.abo0718
- Imai M., Ito M., Kiso M., Yamayoshi S., Uraki R., Fukushi S., et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med. 2023; 388(1): 89–91. https://doi.org/10.1056/NEJMc2214302
- Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022; 602(7898): 657–63. https://doi.org/10.1038/s41586-021-04385-3
- Wang Q., Guo Y., Iketani S., Nair M.S., Li Z., Mohri H., et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature. 2022; 608(7923): 603–8. https://doi.org/10.1038/s41586-022-05053-w
- Pochtovyi A.A., Kustova D.D., Siniavin A.E., Dolzhikova I.V., Shidlovskaya E.V., Shpakova O.G., et al. In vitro efficacy of antivirals and monoclonal antibodies against SARS-CoV-2 Omicron lineages XBB.1.9.1, XBB.1.9.3, XBB.1.5, XBB.1.16, XBB.2.4, BQ.1.1.45, CH.1.1, and CL.1. Vaccines (Basel). 2023; 11(10): 1533. https://doi.org/10.3390/vaccines11101533
- Puhach O., Meyer B., Eckerle I. SARS-CoV-2 viral load and shedding kinetics. Nat. Rev. Microbiol. 2023; 21(3): 147–61. https://doi.org/10.1038/s41579-022-00822-w
- Gushchin V.A., Pochtovyi A.A., Kustova D.D., Ogarkova D.A., Tarnovetskii I.Y., Belyaeva E.D., et al. Dynamics of SARS-CoV-2 major genetic lineages in moscow in the context of vaccine prophylaxis. Int. J. Mol. Sci. 2022; 23(23): 14670. https://doi.org/10.3390/ijms232314670
- Wickham H., François R., Henry L., Müller K., Vaughan D. dplyr: A Grammar of Data Manipulation. Available at: https://dplyr.tidyverse.org/reference/dplyr-package.html
- Wickham H. ggplot2. New York, NY: Springer; 2009. https://doi.org/10.1007/978-0-387-98141-3
- Patil I. Visualizations with statistical details: The “ggstatsplot” approach. J. Open. Source. Softw. 2021; 6(61): 3167. https://doi.org/10.21105/joss.03167
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020; 182(4): 812–27.e19. https://doi.org/10.1016/j.cell.2020.06.043
- Markov P.V., Ghafari M., Beer M., Lythgoe K., Simmonds P., Stilianakis N.I., et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023; 21(6): 361–79. https://doi.org/10.1038/s41579-023-00878-2
- Klink G.V., Safina K.R., Nabieva E., Shvyrev N., Garushyants S., Alekseeva E., et al. The rise and spread of the SARS-CoV-2 AY.122 lineage in Russia. Virus Evol. 2022; 8(1): veac017. https://doi.org/10.1093/ve/veac017
- Teyssou E., Delagrèverie H., Visseaux B., Lambert-Niclot S., Brichler S., Ferre V., et al. The Delta SARS-CoV-2 variant has a higher viral load than the Beta and the historical variants in nasopharyngeal samples from newly diagnosed COVID-19 patients. J. Infect. 2021; 83(4): e1–3. https://doi.org/10.1016/j.jinf.2021.08.027
- von Wintersdorff C.J.H., Dingemans J., van Alphen L.B., Wolffs P.F.G., van der Veer B.M.J.W., Hoebe C.J.P.A., et al. Infections with the SARS-CoV-2 Delta variant exhibit fourfold increased viral loads in the upper airways compared to Alpha or non-variants of concern. Sci. Rep. 2022; 12(1): 13922. https://doi.org/10.1038/s41598-022-18279-5
- Guo H., Jiang J., Shen S., Ge X., Fan Q., Zhou B., et al. Additional mutations based on Omicron BA.2.75 mediate its further evasion from broadly neutralizing antibodies. IScience. 2023; 26(4): 106283. https://doi.org/10.1016/j.isci.2023.106283
- Motozono C., Toyoda M., Zahradnik J., Saito A., Nasser H., Tan T.S., et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021; 29(7): 1124–36.e11. https://doi.org/10.1016/j.chom.2021.06.006
- Singh P., Sharma K., Singh P., Bhargava A., Negi S.S., Sharma P., et al. Genomic characterization unravelling the causative role of SARS-CoV-2 Delta variant of lineage B.1.617.2 in 2nd wave of COVID-19 pandemic in Chhattisgarh, India. Microb. Pathog. 2022; 164: 105404. https://doi.org/10.1016/j.micpath.2022.105404
- Sarkar P., Banerjee S., Saha S.A., Mitra P., Sarkar S. Genome surveillance of SARS-CoV-2 variants and their role in pathogenesis focusing on second wave of COVID-19 in India. Sci. Rep. 2023; 13(1): 4692. https://doi.org/10.1038/s41598-023-30815-5
- Lin X., Sha Z., Trimpert J., Kunec D., Jiang C., Xiong Y., et al. The NSP4 T492I mutation increases SARS-CoV-2 infectivity by altering non-structural protein cleavage. Cell Host Microbe. 2023; 31(7): 1170–84. https://doi.org/10.1016/j.chom.2023.06.002
- Kim S.M., Kim E.H., Anthony M., Casel B., Kim Y.I., Sun R., et al. SARS-CoV-2 variants show temperature-dependent enhanced polymerase activity in the upper respiratory tract and high transmissibility. bioRxiv. 2022. Preprint. https://doi.org/10.1101/2022.09.27.509689.
- Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., Batra N., et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Euro Surveill. 2021; 26(24): 2100509. https://doi.org/10.2807/1560-7917.ES.2021.26.24.2100509
- Earnest R., Uddin R., Matluk N., Renzette N., Turbett S.E., Siddle K.J., et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. Cell Rep. Med. 2022; 3(4): 100583. https://doi.org/10.1016/j.xcrm.2022.100583
- Fan Y., Li X., Zhang L., Wan S., Zhang L., Zhou F. SARS-CoV-2 Omicron variant: recent progress and future perspectives. Signal Transduct. Target. Ther. 2022; 7(1): 141. https://doi.org/10.1038/s41392-022-00997-x
- Sukhikh G.T., Priputnevich T.V., Ogarkova D.A., Pochtovyi A.A., Kustova D.D., Zlobin V.I., et al. Sputnik light and Sputnik V vaccination is effective at protecting medical personnel from COVID-19 during the period of Delta variant dominance. Vaccines (Basel). 2022; 10(11): 1804. https://doi.org/10.3390/vaccines10111804
- Planas D., Veyer D., Baidaliuk A., Staropoli I., Guivel-Benhassine F., Rajah M.M., et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021; 596(7871): 276–80. https://doi.org/10.1038/s41586-021-03777-9
- Simon-Loriere E., Schwartz O. Towards SARS-CoV-2 serotypes? Nat. Rev. Microbiol. 2022; 20(4): 187–8. https://doi.org/10.1038/s41579-022-00708-x
- Wu H., Xing N., Meng K., Fu B., Xue W., Dong P., et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021; 29(12): 1788–801. https://doi.org/10.1016/j.chom.2021.11.005
- Bills C.J., Xia H., Chen J.Y.C., Yeung J., Kalveram B.K., Walker D., et al. Mutations in SARS-CoV-2 variant nsp6 enhance type-I interferon antagonism. Emerg. Microbes Infect. 2023; 12(1): 2209208. https://doi.org/10.1080/22221751.2023.2209208
- Liu Y., Liu J., Plante K.S., Plante J.A., Xie X., Zhang X., et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022; 602(7896): 294–99. https://doi.org/10.1038/s41586-021-04245-0
- Escalera A., Gonzalez-Reiche A.S., Aslam S., Mena I., Laporte M., Pearl R.L., et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe. 2022; 30(3): 373–87.e7. https://doi.org/10.1016/j.chom.2022.01.006
- Starr T.N., Greaney A.J., Hannon W.W., Loes A.N., Hauser K., Dillen J.R., et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science. 2022; 377(6604): 420–4. https://doi.org/10.1126/science.abo7896
- Hirotsu Y., Maejima M., Shibusawa M., Natori Y., Nagakubo Y., Hosaka K., et al. SARS-CoV-2 Omicron sublineage BA.2 replaces BA.1.1: Genomic surveillance in Japan from September 2021 to March 2022. J. Infect. 2022; 85(2): 174–211. https://doi.org/10.1016/j.jinf.2022.04.040
- Mastrorosa I., Cozzi-Lepri A., Colavita F., Lalle E., Mazzotta V., Cimaglia C., et al. SARS-CoV-2 nasopharyngeal viral load in individuals infected with BA.2, compared to Alpha, Gamma, Delta and BA.1 variants: A single-center comparative analysis. J. Clin. Virol. 2022; 157: 105299. https://doi.org/10.1016/j.jcv.2022.105299
- Tozer K., Sjaarda C.P., Moslinger E., Wong H., Mubareka S., Maguire F., et al. Comparison of SARS-CoV-2 viral loads in the nasal mucosa of patients infected with BA.1, BA.2, or BA.5 Omicron lineages. Open Forum Infect. Dis. 2022; 9(12): ofac564. https://doi.org/10.1093/ofid/ofac564
- Takatsuki Y., Takahashi Y., Nakajima J., Iwasaki Y., Nagano K., Tani-Sassa C., et al. Viral load of SARS-CoV-2 Omicron BA.5 is lower than that of BA.2 despite the higher infectivity of BA.5. Immun. Inflamm. Dis. 2023; 11(2): e783. https://doi.org/10.1002/iid3.783
- Cao Y., Yisimayi A., Jian F., Song W., Xiao T., Wang L., et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022; 608(7923): 593–602. https://doi.org/10.1038/s41586-022-04980-y
- Tuekprakhon A., Nutalai R., Dijokaite-Guraliuc A., Zhou D., Ginn H.M., Selvaraj M., et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell. 2022; 185(14): 2422–33. https://doi.org/10.1016/j.cell.2022.06.005
- Rashid F., Xie Z., Suleman M., Shah A., Khan S., Luo S. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 2022; 13: 940756. https://doi.org/10.3389/fimmu.2022.940756
- Github. BA.5.1.29 sublineage with S:K150E, S:460K, Orf8:F41C. Available at: https://github.com/cov-lineages/pango-designation/issues/1187
- Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., et al. GISAID’s role in pandemic response. China CDC Wkly. 2021; 3(49): 1049–51. https://doi.org/10.46234/ccdcw2021.255
- Haslwanter D., Dieterle M.E., Wec A.Z., O’Brien C.M., Sakharkar M., Florez C., et al. A combination of receptor-binding domain and N-terminal domain neutralizing antibodies limits the generation of SARS-CoV-2 spike neutralization-escape mutants. mBio. 2021; 12(5): e0247321. https://doi.org/10.1128/mBio.02473-21
- Kee J., Thudium S., Renner D.M., Glastad K., Palozola K., Zhang Z., et al. SARS-CoV-2 disrupts host epigenetic regulation via histone mimicry. Nature. 2022; 610(7931): 381–8. https://doi.org/10.1038/s41586-022-05282-z
- Hossain A., Akter S., Rashid A.A., Khair S., Alam A.S.M.R.U. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathog. 2022; 170: 105699. https://doi.org/10.1016/j.micpath.2022.105699
- Github. BJ.1/BM.1.1.1 (=BA.2.75.3.1.1.1) recombinant with breakpoint in S1 (>=5 sequences, 3x Singapore, 2x US as of 2022-09-12). Available at: https://github.com/cov-lineages/pango-designation/issues/1058
- Tamura T., Ito J., Uriu K., Zahradnik J., Kida I., Anraku Y., et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023; 14(1): 2800. https://doi.org/10.1038/s41467-023-38435-3
- Ao D., He X., Hong W., Wei X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm. (2020). 2023; 4(2): e239. https://doi.org/10.1002/mco2.239
- Parums D.V. Editorial: The XBB.1.5 (‘Kraken’) subvariant of Omicron SARS-CoV-2 and its rapid global spread. Med. Sci. Monitor. 2023; 29: e939580. https://doi.org/10.12659/MSM.939580
Дополнительные файлы

