



The role of the encephalomyocarditis virus type 1 proteins L and 2A in the inhibition of the synthesis of cellular proteins and the accumulation of viral proteins during infection
- Authors: Ivin Y.Y.1, Butusova A.A.1, Gladneva E.E.1, Kolomijtseva G.Y.2, Khapchaev Y.K.1, Ishmukhametov A.A.1,3
-
Affiliations:
- Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute)
- A.N. Belozersky Research Institute of Physico-Chemical Biology MSU
- Institute for Translational Medicine and Biotechnology, Sechenov First Moscow State Medical University
- Issue: Vol 68, No 5 (2023)
- Pages: 428-444
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/446
- DOI: https://doi.org/10.36233/0507-4088-195
- EDN: https://elibrary.ru/eeiuhq
- ID: 446

Cite item
Full Text

Abstract
Introduction. Infection of cells with encephalomyocarditis virus type 1 (EMCV-1, Cardiovirus A: Picornaviridae) is accompanied by suppression of cellular protein synthesis. The main role in the inhibition of cellular translation is assigned to the L and 2A «security» proteins. The mechanism of the possible influence of the L protein on cellular translation is unknown. There are hypotheses about the mechanism of influence of 2A protein on the efficiency of cap-dependent translation, which are based on interaction with translation factors and ribosome subunits. However, the available experimental data are contradictory, obtained using different approaches, and do not form a unified model of the interaction between the L and 2A proteins and the cellular translation machinery.
Aim. To study the role of L and 2A «security» proteins in the suppression of translation of cellular proteins and the efficiency of translation and processing of viral proteins in infected cells.
Materials and methods. Mutant variants of EMCV-1 were obtained to study the properties of L and 2A viral proteins: Zfmut, which has a defective L; Δ2A encoding a partially deleted 2A; Zfmut&Δ2A containing mutations in both proteins. Translational processes in infected cells were studied by Western-blot and the pulse method of incorporating radioactively labeled amino acids (14C) into newly synthesized proteins, followed by radioautography.
Results. The functional inactivation of the 2A protein does not affect the inhibition of cellular protein synthesis. A direct correlation was found between the presence of active L protein and specific inactivation of cellular protein synthesis at an early stage of viral infection. Nonspecific suppression of the translational processes of the infected cell, accompanied by phosphorylation of eIF2α, occurs at the late stage of infection. Partial removal of the 2A protein from the EMCV-1 genome does not affect the development of this process, while inactivation of the L protein accelerates the onset of complete inhibition of protein synthesis. Partial deletion of the 2A disrupts the processing of viral capsid proteins. Suppression of L protein functions leads to a decrease in the efficiency of viral translation.
Conclusion. A study of the role of EMCV-1 L and 2A proteins during the translational processes of an infected cell, first performed using infectious viral pathogens lacking active L and 2A proteins in one experiment, showed that 2A protein is not implicated in the inhibition of cellular translation in HeLa cells; L protein seems to play an important role not only in the specific inhibition of cellular translation but also in maintaining the efficient synthesis of viral proteins; 2A protein is involved not only in primary but also in secondary processing of EMCV-1 capsid proteins.
Full Text
Введение
Вирусы являются внутриклеточными паразитами, которые используют энергетические и синтетические системы клетки-хозяина для осуществления собственного размножения. Не является исключением также и клеточная система синтеза белков. В ходе развития вирусных инфекций в клеточных линиях и организмах накопление вирусных структурных и неструктурных белков осуществляется за счет использования клеточных рибосом. Как правило, при этом трансляционный аппарат хозяина подвергается воздействию со стороны вирусов. Более того, данное воздействие может быть использовано вирусным агентом для контроля синтеза клеточных белков с целью ограничения работы систем врожденного иммунитета. Серотип вируса энцефаломиокардита (EMCV) типа 1 (EMCV-1) принадлежит к виду Cardiovirus A рода Cardiovirus семейства Picornaviridae1. Род включает в себя несколько представителей вирусов позвоночных животных: EMCV, вирус энцефаломиелита мышей Тейлера (TMEV), кардиовирусы полевок, а также два представителя вирусов человека: вирус Саффолд (SAFV) и вирус вилюйского энцефалита (VHEV), который в последние годы идентифицировали как представителя этого рода [1]. Известно, что SAFV может поражать нервную, сердечно-сосудистую и эндокринную системы человека и вызывать энтерит, миокардит и, предположительно, рассеянный склероз [2, 3]. VHEV изучен слабо, однако исследователи предполагают, что он вызывает тяжелые нейродегенеративные заболевания человека [4]. Вирусы EMCV и TMEV являются модельными представителями рода, широкое и всестороннее изучение которых поможет в понимании функционирования остальных представителей. Серотипы EMCV инфицируют множество позвоночных животных разных отрядов; более того, описаны случаи заражения человека [5], что делает этот модельный вирус еще более привлекательным для изучения. EMCV имеет икосаэдрический капсид диаметром 27–30 нм и геном размером 7,5 т.н., который кодирует полипротеин, подвергающийся постадийному процессингу на 12 отдельных белков, один дополнительный белок кодируется в альтернативной рамке считывания [5]. Геном EMCV имеет структурированный участок РНК в 5’-нетранслируемой области, который ответственен за связывание с рибосомальным комплексом, – внутренний участок посадки рибосом (IRES) II типа [5]. Он позволяет протекать процессу синтеза вирусного полипротеина по иному механизму, чем тот, по которому синтезируется подавляющее большинство белков клеток-хозяина.
Классический кэп-зависимый механизм инициации трансляции эукариот заключается в том, что узнавание клеточной мРНК и вовлечение ее в инициаторный комплекс 43S/мРНК происходит благодаря связыванию 5’-концевой структуры кэпа m7G с комплексом eIF4F. Этот комплекс состоит из кэп-связывающего фактора eIF4E, РНК-хеликазы eIF4A и белка eIF4G, связывающего несколько факторов и формирующего мостик между РНК и рибосомой. Помимо этого, для инициации трансляции и связывания мРНК необходимо привлечение eIF4B, eIF4H и поли-А-связывающего белка (PABP). Формирование комплекса 43S, второй части инициаторного комплекса 43S/мРНК, проходит путем ассоциации малой субъединицы рибосомы 40S c факторами eIF1, eIF1A, eIF3, eIF5, а также важнейшего фактора eIF2, переносящего комплекс инициаторной аминокислоты Мет-тРНК [6]. Гуанозинтрифосфат (GTP), связанный с eIF2-Мет-тРНК, гидролизуется после формирования комплекса 43S, и комплекс eIF2 с гуанозиндифосфатом (GDP) высвобождается. Для дальнейшего связывания eIF2-GDP с Мет-тРНК и начала нового раунда инициации трансляции необходим обмен GDP на GTP, который осуществляет белок eIF2B [7]. Один из способов контроля уровня синтеза белков заключается в изменении статуса фосфорилирования субъединицы α фактора eIF2 по остатку серина-57. Фосфорилированная форма eIF2-GDP (eIF2α-P) превращается из субстрата eIF2B в ингибитор, таким образом содержание активированного комплекса eIF2-GTP резко сокращается [7]. В эукариотических клетках фосфорилирование eIF2α осуществляют четыре известные киназы, активирующиеся при различных видах стресса или сигнала: PKR, PERK, GCN2 и HRI [8, 9].
Для инициации белкового синтеза по IRES-зависимому механизму с участием пикорнавирусной РНК, лишенной кэп-структуры m7G, кэп-связывающий фактор eIF4E не является необходимым [10]. Более того, для инициации синтеза пикорнавирусных белков не является необходимым связывание поли-А-хвоста генома с PABP и формирование кольцевой структуры 43S/RNA. Существуют четыре известных типов IRES, различающиеся по структуре и клеточным факторам инициации, необходимым для формирования инициаторного комплекса. Наиболее распространенные типы IRES пикорнавирусов I (представители рода Enterovirus) и II (представители родов Aphtovirus и Cardiovirus) нуждаются в факторах eIF1, eIF1A, eIF2, eIF3, eIF5, eIF4A, eIF4B [11]. Также необходимым является наличие С-концевой части eIF4G, ответственной за связывание eIF3, eIF4A, eIF4B и вирусной (+) РНК. Избирательное подавление синтеза клеточных белков в ходе инфекции становится возможным благодаря подобному альтернативному механизму инициации трансляции [12, 13].
Необходимо отметить, что клеточные механизмы контроля трансляции могут управлять и синтезом вирусных белков, являясь частью противовирусного иммунитета. К примеру, фосфорилирование eIF2α, блокирует синтез не только клеточных белков, но и белков EMCV-1 [14].
Известные механизмы избирательного подавления клеточной трансляции при пикорнавирусной инфекции основываются на различии в необходимом наборе инициаторных факторов для кэп- и IRES-зависимой инициации трансляции. Эти механизмы, как правило, связаны с активностью белков L и 2А. Они входят в особую разнообразную группу неструктурных белков, называемых «секьюрити»-белками или факторами вирулентности. В полипротеине L и 2А фланкируют белки капсида [15]. Так, протеазы 2Аpro представителей рода Enterovirus и Lpro рода Aphtovirus расщепляют фактор инициации eIF4G, отделяя N-конец, который содержит сайты связывания eIF4E и PABP, и лишая возможности клеточным мРНК ассоциироваться с eIF4F посредством структуры кэп m7G и поли-А-хвоста [16–18]. Более того, белок 2Аpro энтеровирусов в ходе инфекции вызывает деградацию PABP [19]. Этот набор реакций в ходе инфекции приводит к избирательному подавлению кэп-зависимой трансляции. Среди «секьюрити»-белков представителей рода Cardiovirus отсутствуют указанные протеазы, у L и 2А не обнаружены какие-либо виды протеолитической активности, однако подавление трансляции клетки-хозяина при этом имеет место [20]. Это подавление не связано с расщеплением факторов инициации eIF4G и PABP, что свидетельствует об альтернативном механизме ингибирования синтеза клеточных белков. Однако роль в ингибировании приписывают также «секьюрити»-белкам L и 2А [21, 22].
Лидерный белок L EMCV состоит из 67 аминокислотных остатков и имеет в составе функциональный домен Zn-палец [23], связывающий ион Zn2+ в качестве лиганда, и кислотный домен, подвергающийся фосфорилированию клеточными киназами [24]. Показано, что домен Zn-палец ответственен за подавление апоптотической программы в клетках HeLa [25], принимает участие в фосфорилировании нуклеопоринов и нарушении ядерно-цитоплазматического транспорта [26], в подавлении синтеза интерферонов-α/β [27].
Белки 2А представителей рода Cardiovirus, и EMCV в частности, также имеют несколько функциональных доменов. На границе между белками 2А и 2B расположен мотив NPG(P), по которому происходит котрансляционный разрыв полипептидной цепи, что играет ключевую роль в первичном процессинге полипротеина – в ходе синтеза происходит отделение предшественника L-P1-2A от остальных неструктурных белков [28]. В ранних исследованиях в составе белка 2А был предсказан сигнал ядерной локализации (с 94 по 102 а.о.) [21, 29]. Показано, что белок направляется в ядро и данный мотив играет, по-видимому, решающую роль. Однако недавно было обнаружено, что этот участок играет ключевую роль в функционировании белка 2А как трансактиватора −1 рибосомального сдвига при синтезе белков кардиовирусов [30]. Участок с 95 по 100 а.о. (для EMCV), названный аргининовой петлей, связывается с РНК-шпилькой, формирующейся в 5’-концевой области 2B-кодирующего фрагмента генома. Связывание белка 2А со шпилькой вызывает котрасляционный рибосомальный сдвиг рамки считывания в так называемом «скользком» сайте, который расположен перед шпилькой. Рибосомальный сдвиг вызывает образование белка 2B*, на котором заканчивается синтез полипептидной цепи. Показано, что запуск рибосомального сдвига коррелирует с накоплением белка 2А в клетке и происходит через 4–8 ч после начала инфекции. Таким образом, белок 2А функционирует как переключатель уровня синтеза белков, которые располагаются на 3’-конце генома [30–33].
Существует множество гипотез и свидетельств вероятного участия белков L и 2А EMCV в модификации трансляционного аппарата клеток. Эксперименты в клеточных линиях BHK-21 и L929 с вирусом с частично делетированным белком L указывали на возможное участие этого белка в ингибировании клеточной трансляции: в отсутствие белка L была обнаружена задержка процесса подавления клеточного белкового синтеза [22]. Белку 2А посвящено большое число исследований, касающихся изучения его возможной роли в ингибировании клеточной трансляции. Подтверждающие данные были обнаружены в экспериментах как in vitro (на модели изучения белкового синтеза с использованием репликонных конструкций [21]), так и in vivo (на вирусной модели в культуре клеток BHK-21 [34]). В обоих случаях исследователи вносили делеции в последовательность белка 2А. Разработаны несколько моделей, которые пытаются объяснить возможную роль 2А в этом процессе. Одна из них основана на нарушении функционирования фактора инициации eIF4E, который не является необходимым для IRES-зависимой трансляции EMCV [11], причем предполагается несколько возможных сценариев влияния. По одному из них белок 2А может влиять на уровень фосфорилирования 4E-BP, что приводит к ингибированию eIF4E. Данный феномен был обнаружен в экспериментах в культуре клеток BHK [34], но не был найден в других культурах [29]. По другому сценарию предполагается, что белок 2А напрямую связывается с eIF4E посредством С-концевого мотива, содержащего аминокислотные остатки с 126-го по 134-й, который аналогичен по составу с eIF4E-связывающим доменом 4E-BP [29]. Авторы предсказали указанный сайт связывания, а также получили результаты in vitro взаимодействия 2А и eIF4E, которое нарушалось при введении мутаций в потенциальный сайт связывания. Однако эксперименты с инфицированием клеток мутантными EMCV по предсказанному сайту показали отсутствие эффекта на клеточную трансляцию [29]. Более того, последние данные о структурных особенностях белка 2А продемонстрировали отсутствие гомологии между предполагаемым сайтом связывания белка 2А с eIF4E и таковым в составе белка 4E-BP [31, 33], что ставит под сомнение легитимность этой модели.
Альтернативная модель, объясняющая возможную роль 2А в процессе ингибирования клеточной трансляции, основана на взаимодействии белка 2А с рибосомальным комплексом. Потенциальное взаимодействие с субъединицами рибосом было установлено еще несколько десятилетий назад [35, 36], однако в последние годы эти данные были подтверждены кристаллографическими исследованиями, в которых выяснили, что белок 2А взаимодействует с рибосомальной РНК посредством РНК-связывающего домена, который участвует во взаимодействие со шпилькой при активации −1 рибосомального сдвига [31, 33]. Данная модель не объясняет механизм специфичного подавления клеточной трансляции.
Несмотря на обилие гипотез, которые могут объяснить вероятную роль белка 2А в подавлении клеточной трансляции, результаты разных исследователей противоречат друг другу. Механизм возможного влияния белка L на трансляционные процессы также остается неизвестным.
Цель исследования заключалась в уточнении роли «секьюрити»-белков L и 2А в подавлении трансляции клеточных белков, которая наблюдается при инфекции EMCV-1, а также роли этих белков в трансляции вирусного генома. Для этого мы инактивировали оба белка L и 2А в геноме штамма Mengo EMCV-1, получив 3 мутантных вируса: два вируса имели мутацию по одному из белков; один – лишенный активных «секьюрити»-белков, был получен впервые. Такой подход позволяет изучать влияние этих белков в одних и тех же стандартных и контролируемых условиях.
Материалы и методы
Вирусы и клеточные культуры
В работе использовали клеточные культуры HeLa-B и BHK-21. Клетки культивировали в питательной среде ДМЕМ, содержащей 10% эмбриональной бычьей сыворотки (Gibco), при температуре 37 °C в атмосфере, содержащей 5% CO2. Вирус EMCV-1 штамма Mengo дикого типа (далее – WT) был получен путем трансфекции клеток BHK-21 полногеномной вирусной РНК, образовавшейся после in vitro транскрипции плазмидной ДНК pM16.1 [37]. Перед использованием в экспериментах WT был дважды пассирован в культуре клеток BHK-21. Мутантный вариант вируса EMCV-1 Zfmut, кодирующий белок L с двумя аминокислотными заменами Cys19/Ala и Cys22/Ala, которые разрушают функциональный домен Zn-палец, был описан ранее и, как WT, получен из плазмидной ДНК pM16.1Zfmut [25, 38]. Перед использованием в экспериментах мутантный вариант EMCV-1 Zfmut был дважды пассирован в культуре клеток BHK-21. Мутантные варианты вируса EMCV-1 Δ2A и Zfmut&Δ2A были получены аналогичным образом из конструкций pM16.2Δ2A и pM16.1Zfmut&Δ2A соответственно. Перед использованием мутантные варианты Δ2A и Zfmut&Δ2A прошли последовательные три пассажа в культуре клеток BHK-21, процедуру клонирования методом негативных колоний и два дополнительных пассажа после клонирования. Вирус Δ2A кодирует частично делетированный белок 2А (с 11 по 125 а.о.), Zfmut&Δ2A имеет описанные мутации в обоих белках L и 2А.
Плазмидные конструкции
pM16.1 содержит полногеномную копию штамма Mengo серотипа EMCV-1 [37]. Конструкция pM16.1Zfmut содержит полногеномную копию штамма Mengo, имеющего две аминокислотные замены Cys19/Ala и Cys22/Ala в последовательности белка L [27]. Конструкция pM16.1Δ2A кодирует полногеномную копию штамма Mengo, имеющего делецию длиной 115 а.о. в последовательности белка 2А (с 11 по 125 а.о.). Конструкция была получена в рамках настоящей работы на основе pM16.1. Для этого применяли прием «перекрывающейся» полимеразной цепной реакции (ПЦР): 1) амплификация двух фрагментов, кодирующих N-концевой и С-концевой фрагменты 2А с помощью соответствующих пар олигонуклеотидов («Синтол», Москва): Δ2A_1 (5’- CTCCCAAGCAAAGCAGCG -3’) – Δ2A_2 (5’- TATGCAGGATACTTTTCAGATC -3’) и Δ2A_3 (5’- CTGAAAAGTATCCTGCATAAGTTTTAGAGACATCCAAAGGG -3’) – Δ2A_4 (5’- TCAAGACACAACCACTTGCC -3’); 2) лигирование получившихся двух ампликонов с помощью ПЦР благодаря комплиментарным участкам с помощью концевых олигонуклеотидов Δ2A_1 и Δ2A_4. Полученный фрагмент, кодирующий делетированный белок 2А, был встроен в pM16.1 с помощью обработки эндонуклеазами рестрикции AflII и SacII и последующим лигированием с помощью лигазы T4 (ThermoFisher Scientific, Литва). Конструкция pM16.1Zfmut&Δ2A кодирует полногеномную копию штамма Mengo, имеющего описанные ранее модификации в белке L и 2А одновременно, была сконструирована аналогично на основе конструкции pM16.1Zfmut.
In vitro транскрипция и трансфекция
Плазмидные ДНК pM16.1, pM16.1Zfmut, pM16.1Δ2A и pM16.1Zfmut&Δ2A были линеаризованы с помощью эндонуклеазы рестрикции BamHI и очищены с помощью набора QIAquick (Qiagen, США). Геномная РНК была транскрибирована с использованием РНК-полимеразы Т7 (ThermoFisher Scientific, Литва). Полученные РНК трансфицировали в клетки BHK-21 c помощью DEAE-декстранового метода (Promega, США). Клетки инкубировали при температуре 37 °C до деградации клеточного монослоя. Содержимое флаконов подвергали двукратной процедуре заморозки/разморозки при температуре −70 °C.
Пассирование вирусов, клонирование и титрование методом негативных колоний
Используемые вирусы пассировали в культуре клеток BHK-21 при температуре 37 °C в течение 1–2 сут до полного разрушения монослоя. Доза заражения культуры клеток при проведении пассажей составляла от 0,1 до 1 бляшкообразующей единицы (БОЕ) на клетку. Титрование вирусов проводили с применением метода негативных колоний («бляшек») в культуре клеток BHK-21 по ранее описанному протоколу [25]. Для клонирования вирусов, направленного на получение гомогенизированных пулов мутантов, использовали аналогичную процедуру, где вместо фиксации клетки прижизненно окрашивали раствором красителя нейтрального красного (0,02% в солевом растворе Эрла). Вирусные клоны из образованных негативных колоний отбирали и использовали для последующей работы.
Определение урожая вирусов
Клетки HeLa культивировали в 6-луночных планшетах в течение суток до достижения 70–80% конфлюэнтности монослоя. Клетки из нескольких лунок диспергировали с помощью растворов Версена и трипсина и проводили подсчет клеток с помощью цитометра. Перед заражением клеток среду роста удаляли, лунки промывали трижды средой ДМЕМ без сыворотки. В лунки наносили по 300 мкл вирусной суспензии с расчетом множественности заражения 40 БОЕ/кл и инкубировали клетки при комнатной температуре (КТ) в течение 30 мин. Лунки промывали дважды и добавляли по 1 мл среды ДМЕМ без сыворотки. После инкубации при температуре 37 °C в течение необходимого времени проводили процедуру двукратной заморозки/разморозки среды с клетками и анализировали полученную суспензию титрованием методом негативных колоний.
Изучение белкового синтеза. Пульс-метод
Клетки HeLa культивировали в 12-луночных планшетах в течение суток до достижения 70–80% конфлюэнтности монослоя. Заражение клеток проводили с расчетом множественности заражения 40 БОЕ/кл. Внесение радиоактивной метки и получение радиоавтографов осуществляли по методике, описанной ранее [38].
Для оценки включения метки в новосинтезированные белки на разных стадиях инфицирования использовали сцинтилляционный счетчик. Для этого выровненные по содержанию β-актина лизаты клеток наносили на мембраны из фильтровальной бумаги. Мембраны высушивали и отмывали от невстроенных меченых аминокислот кипячением в течение 5 мин в растворе 10% уксусной кислоты и 20% этанола и последующим промыванием в 96% этаноле. Высушенные мембраны помещали в пробирки и обрабатывали 10 мл коктейля солюбилизатора и сцинтиллятора Ultima Gold F (Perkin Elmer). После инкубации в течение 2 ч измеряли импульсы на сцинтилляционном счетчике. Полученные данные нормировали по показаниям встройки радиоактивно меченых аминокислот в белки незараженной культуры клеток.
Вестерн-блот-гибридизация
Клеточные лизаты, полученные в ходе экспериментов по анализу белкового синтеза, анализировали методом вестерн-блот-гибридизации для оценки содержания β-актина, фосфорилированной формы eIF2α и вирусных белков. Для этого образцы разделяли в 12% полиакриламидном геле, переносили белковые компоненты на нитроцеллюлозную мембрану. Мембрану блокировали с помощью 5% раствора сухого обезжиренного молока в трис-буферном растворе (TBS-T, 50 мМ TRIS рН 7,6, 150 мМ NaCl, 0,05% Tween-20) в течение 1 ч и гибридизовали с необходимыми антителами или поликлональной сывороткой в описанном растворе сухого молока. Все процедуры проводили при КТ. Для детекции β-актина использовали антитела, конъюгированные с пероксидазой хрена (HRP) (Sigma, A5316, разведение 1 : 25 000). Для анализа уровня фосфорилирования eIF2α применяли комбинацию из первичных кроличьих антител против p-eIF2α (pSer51, разведение 1 : 1000, Sigma) и вторичных антител против иммуноглобулинов кролика, конъюгированных с HRP (разведение 1 : 2500, Promega). Для детекции вирусных белков EMCV-1 использовали комбинацию из поликлональной мышиной сыворотки, полученной ранее в лаборатории, и вторичных антител против иммуноглобулинов мыши, конъюгированных с HRP (разведение 1 : 2500, Promega). Результаты гибридизации визуализировали с помощью набора ECL-Plus (Promega) и рентгеновской пленки (XBM x-ray Retina blue sensitive). Анализ изображений иммуноблотов проводили с помощью программы ImageJ.
Количественный ПЦР в режиме реального времени
Для оценки количества вирусной геномной РНК использовали метод количественного ПЦР в режиме реального времени (qPCR). Клетки для эксперимента выращивали в 12-луночных планшетах и инфицировали, как указано ранее. Процедуру проводили по методике, опубликованной ранее, с использованием олигонуклеотидов: MGVL1 (5’- CGCTAGGAATGCGTAGAACA -3’), MGVR1 (5’- AGCTCGTCCTTGAGGAATGT -3’), MGVP1 (5’- 6-FAM-TGGGAAACCGCCACTCTTATCCC-BHQ1 -3’) в качестве флуоресцентной пробы [25].
Результаты
Получение и фенотипическая характеристика мутантных вирусов с делецией в белке 2А
Для проведения исследований использовали вирусные пулы WT и Zfmut, которые прошли 3 пассажа на культуре клеток BHK-21 и были подробно охарактеризованы ранее [25, 26, 38].
Мутантные вирусы Δ2A и Zfmut&Δ2A, имеющие частично делетированный белок 2А (рис. 1 а), в первом пассаже после трансфекции имели неоднородный фенотип негативных колоний, что может свидетельствовать о генетической неоднородности полученных пулов. Для достижения гомогенной популяции по фенотипу проводили дополнительные пассажи в культуре клеток BHK-21 (рис. 1 б). Необходимо отметить, что оба полученных вируса в процессе пассирования адаптировались к культуре, средний размер негативных колоний увеличился, они стали более гомогенными по размеру. Для отбора генетически однородной популяции вирусы Δ2A и Zfmut&Δ2A после 4 пассажей клонировали методом негативных колоний на монослойной культуре клеток BHK-21. Отдельные клоны размножали для получения рабочих пулов на культуре клеток BHK-21 в течение 2 пассажей.

Рис. 1. Схема мутаций, внесенных в последовательность белков L и 2А вируса EMCV-1 для получения мутантных вирусов Zfmut, Δ2A и Zfmut&Δ2A (а); изменение фенотипа негативных колоний полученных мутантных вирусов Δ2A и Zfmut&Δ2A в ходе проведе- ния пассажей на культуре клеток BHK-21 (б).
Fig. 1. Scheme of mutations introduced into the sequence of L and 2A proteins of the EMCV-1 to obtain mutant viruses Zfmut, Δ2A and Zfmut&Δ2A (a); Changes in the plaque phenotype of the obtained mutant viruses Δ2A and Zfmut&Δ2A during passages on BHK-21 cell culture (b).
Полногеномное секвенирование полученных пулов вирусов по методу Сэнгера подтвердило наличие искомой делеции у мутантных вирусов Δ2A и Zfmut&Δ2A, а также выявило аминокислотную замену в положении 99 белка VP2 (VP2Thr99/Ala). Данная мутация была обнаружена во всех клонах вирусов Δ2A и Zfmut&Δ2A. Секвенирование мутантных вирусов разных пассажей до клонирования выявило постепенное вытеснение из популяции Thr99 на Ala99. Секвенирование WT и Zfmut показало появление данной мутации в части популяции, начиная с 4-го пассажа в клетках BHK-21.

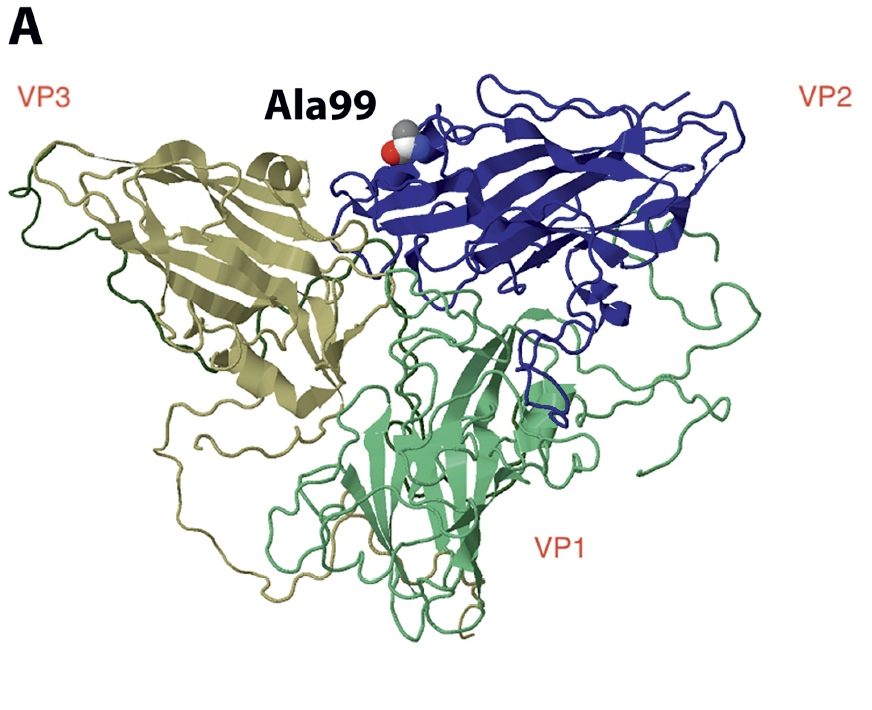
Рис. 2. Положение аминокислотного остатка в позиции № 99 белка VP2 (а); сравнение циклов репродукции EMCV-1 дикого типа (WT) и мутантного вируса с аминокислотной заменой треонина на аланин в положении 99 белка VP2 (Thr/VP2_99/Ala) (б).
Fig. 2. The location of the amino acid residue at position 99 of the VP2 protein (a). Comparison of the reproduction cycles of wild-type EMCV-1 (WT) and mutant virus with the amino acid substitution of threonine for alanine at position 99 of the VP2 protein (Thr/VP2_99/Ala) (b).
Аминокислотный остаток Ala/Thr99 белка VP2 занимает место в выпетливании полипептидной цепи на границе двух капсомеров, состоящих из белков VP1, VP2 и VP3 (рис. 2 а). Для характеристики влияния обнаруженной мутации был получен вирус EMCV дикого типа с заменой Thr99/Ala в белке VP2 (VP2 Thr99Ala). Мы не обнаружили изменений в репродуктивном цикле (рис. 2 б) и фенотипе негативных колоний (данные не приведены) по сравнению с вирусом дикого типа (WT). Биоинформатический анализ геномов EMCV показал, что аминокислотный остаток Ala в позиции 99 встречается в подавляющем большинстве изолятов и штаммов, кроме двух последовательностей штамма Mengo (изолят M и Rz-pMwt), на основе которых была получена плазмидная конструкция pM16.1, использованная в работе [38]. По всей вероятности, диким генотипом можно считать VP2_Ala99, а в изоляте M произошла адаптационная замена Ala на Thr. В дальнейшем пассирование вирусов, полученных на основе последовательности изолята M, в культуре клеток BHK-21 приводило к возвращению исходного варианта. Обратная аминокислотная замена происходила быстрее в мутантных вирусах (Δ2A и Zfmut&Δ2A). Таким образом, аминокислотная замена в белке VP2 в полученных мутантных вирусах Δ2A и Zfmut&Δ2A, по-видимому, способствовала повышению их жизнеспособности, но не оказывает влияния на жизнеспособность при введении в WT.
Характеристика репродукции мутантных вирусов в клетках HeLa
Изучение свойств полученных мутантных вирусов Δ2A и Zfmut&Δ2A выявило значимые различия в продукции инфекционных вирусных частиц при заражении клеточной культуры HeLa в сравнении с вирусом WT (рис. 3 а).
Наибольшую способность к репродукции проявлял вирус WT. Через 8 ч после начала инфицирования число инфекционных доз достигало 236,2 ± 95,2 БОЕ на 1 клетку. Инактивация одного из белков L или 2А (мутантные вирусы Zfmut и Δ2A соответственно) приводила приблизительно к 5-кратному снижению количества инфекционных частиц: 46,1 ± 14,7 БОЕ/кл у Zfmut и 73,1 ± 46,9 БОЕ/кл у Δ2A. Лишение вируса обоих функциональных «секьюрити»-белков приводило к более чем 20-кратному снижению урожая вируса до 8,4 ± 4,7 БОЕ/кл.
Количественный анализ содержания вирусной РНК показал, что инактивация белка L привела к снижению накопления геномной РНК EMCV-1 в клетках HeLa. Уровень вирусной РНК в клетках, зараженных Zfmut и Zfmut&Δ2A, оказался существенно ниже через 6 ч после начала инфекции, чем в случае инфекции WT (рис. 3 б). Частичная делеция белка 2А не привела к изменению накопления вирусной РНК, как при интактном белке L, в случае мутанта Δ2A, так и при инактивированном L – Zfmut&Δ2A.
Таким образом, снижение продукции инфекционных частиц Zfmut и Zfmut&Δ2A относительно WT можно объяснить снижением накопления вирусной РНК. Однако снижение репродуктивной способности Δ2A и Zfmut&Δ2A относительно WT и Zfmut соответственно, по-видимому, не связано со сниженной репликацией.
Роль белков L и 2А в подавлении белкового синтеза инфицированных клеток HeLa
Изучение трансляционных процессов в инфицированной культуре проводили с использованием пульс-метода, применяя для детектирования de novo синтезированных белков аминокислоты, меченные изотопом 14C. В качестве негативного контроля использовали клетки, обработанные арсенитом натрия (Ars, 100 нМ) и T-2 токсином (50 нМ), ингибиторами синтеза белков, которые были любезно предоставлены С.Е. Дмитриевым (НИИ ФХБ имени А.Н. Белозерского). В качестве позитивного контроля использовали незараженные клетки (Mock). Метод позволяет оценивать эффективность образования белков в выбранный момент цикла вирусного размножения, регистрируя белковые молекулы, наработанные за 30-минутный промежуток времени.
Заражение клеток HeLa вирусом WT приводило к меняющемуся профилю трансляции белков на разных этапах инфекции. К 3 ч после инфицирования заметно снижение уровня новосинтезированных клеточных белков – снижается их сигнал относительно неинфицированных клеток (рис. 4, 3 ч). Об этом свидетельствует падение общего уровня включения радиоактивно меченных аминокислот в белки, синтезируемые в зараженной клетке, к 3 ч на 40% (рис. 5, WT). Результаты радиоавтографии показывают, что в это время идет синтез вирусспецифичных белков (рис. 4). Учитывая, что синтез вирусных белков вносит вклад в общий уровень трансляции в инфицированной клетке, который мы детектировали с помощью оценки уровня включения меченых аминокислот, синтез клеточных белков ингибируется более чем на 40% относительно незараженной культуры. Более точную оценку уровня ингибирования с помощью этого метода достичь невозможно. Таким образом, на данном этапе инфекции (3 ч) мы наблюдали угнетение синтеза клеточных белков при активном синтезе вирусных. К 4 ч после инфицирования уровень встраивания меченых аминокислот повышается до 80% от уровня незараженных клеток (рис. 5 а). В процессе анализа результатов радиоавтографии синтезированных белков на этом этапе инфекции становится очевидным, что в повышение общей синтетической активности большой вклад вносит трансляция вирусспецифичных белков. Об этом свидетельствует усиление их сигнала на радиоавтографе (рис. 4, WT, 4 ч). Синтез клеточных белков на этом этапе остается более низким, чем в незараженных клетках. Следом за указанным пиком синтетической активности, к 5–6 ч наступает резкое падение как синтеза вирусных белков, так и остаточной клеточной трансляции. Об этом свидетельствует резкое снижение уровня встроенных меченых аминокислот до 10–20% от нормы (рис. 5 а, WT), а также практически полное отсутствие сигналов новосинтезированных белков на радиоавтографе (рис. 4, WT, 5 ч). Следует отметить, что описанное падение синтеза сопровождается заметным повышением уровня фосфорилирования eIF2α по остатку Ser-51, что указывает на запуск каскадов фосфорилирования, направленных на подавление белкового синтеза (рис. 4, 5 б, WT). Таким образом, на поздней стадии инфекции WT наблюдается неспецифичное ингибирование трансляционных процессов в клетках HeLa, связанное с фосфорилированием eIF2α.
Частичная делеция белка 2А в геноме, которая присутствует у мутанта Δ2А, не привела к заметному изменению профиля трансляции в экспериментах с использованием наших методик. Инфицирование клеток HeLa мутантом Δ2A, как и WT, приводило к заметному подавлению клеточной трансляции на 3 и 4 ч, повышению синтеза вирусных белков к 4 ч и полному подавлению трансляции к 5 ч (рис. 4, Δ2A). Финальное подавление синтеза также сопровождалось фосфорилированием eIF2α по остатку Ser-51 (рис. 5 б, Δ2A). График уровня встраивания радиоактивно меченных аминокислот при инфекции Δ2A повторяет таковой при WT-инфекции (рис. 5 а). Таким образом, результаты проведенных экспериментов указывают на то, что белок 2А вируса EMCV-1 не принимает участия в модификации биосинтеза белков в зараженных клетках HeLa.
При инфекции клеток HeLa вирусами, имеющими в составе мутантный белок L (Zfmut и ZfmutΔ2A), профиль белкового синтеза изменялся иначе, чем это происходило в случае инфекции WT или Δ2A. К 3 ч после начала инфекции синтез клеточных белков идет со схожей интенсивностью, что и в незараженных клетках. Об этом свидетельствуют яркие сигналы множества синтезирующихся клеточных белков на радиоавтографе (рис. 4, Zfmut и ZfmutΔ2A, 3 ч) и высокий общий уровень встраивания меченых аминокислот в белки относительно показателей WT и Δ2A (~100%, рис. 5 а). На этой стадии инфекции заметен синтез вирусспецифичных белков (рис. 4, Zfmut и ZfmutΔ2A, отмечены стрелками). Следует отметить, что уровень сигнала вирусных белков Zfmut и ZfmutΔ2A снижен по сравнению с WT и Δ2A. К 4 ч наступает резкое снижение синтетических процессов в клетках, зараженных Zfmut и ZfmutΔ2A, синтез клеточных и вирусных белков ингибируется (рис. 4). Этот процесс сопровождается повышением уровня фосфорилирования eIF2α (рис. 4 и 5 б). Подобное глобальное ингибирование белкового синтеза при инфекции WT и Δ2A происходит на 1 ч позже. Эксперименты продемонстрировали, что в отсутствие активного белка L запуск фосфорилирования eIF2α и, следовательно, ингибирования всего белкового синтеза происходит раньше. К тому же в отсутствие активного белка L (Zfmut и ZfmutΔ2A) не происходит специфичного частичного подавления синтеза клеточных белков на ранней стадии инфекции, как это наблюдается при инфекции WT или Δ2A.
Таким образом, была обнаружена зависимость между наличием активного белка L в составе EMCV и специфичным подавлением синтеза клеточных белков на ранней стадии вирусного цикла (3 ч), не связанным с фосфорилированием eIF2α. Белок 2А на этот процесс влияния не оказывает.
Роль белков L и 2А в накоплении вирусных белков и их процессинге
Для оценки эффективности вирусной трансляции и процессинга структурных вирусных белков мы анализировали паттерн вирусных белков методом иммуноблоттинга с помощью сыворотки против структурных белков EMCV-1 через 4 ч после начала инфекции, когда наблюдается наибольшая эффективность синтеза вирусных белков WT и Δ2A (рис. 4). В качестве стандартного образца, содержащего отдельные структурные вирусные белки, использовали вирус WT рабочего пула, который был очищен методом ультрацентрифугирования в градиенте плотности хлорида цезия (очWT). В составе этого образца обнаружены три отдельных капсидных белка: VP1 – 31 кДа, VP2 – 29 кДа и VP3 – 25 кДа (рис. 6 а, 1). С помощью пульс-метода мы регистрировали эффективность накопления вирусных белков на данном этапе инфекции. Анализ с помощью иммунной сыворотки против белков EMCV-1 позволил обнаружить сигналы, соответствующие вирусным белкам. Расшифровку белкового паттерна проводили с использованием стандартного образца очWT (VP0, VP1, VP2 и VP3), а также по данным литературы [5, 22, 34, 39]. У WT и Zfmut самым большим белком-предшественником после первичного процессинга полипротеина являлся (L)P1-2A массой ~116 кДа, в случае мутантов Δ2A и Zfmut&Δ2A, ожидаемо, предшественник (L)P1-Δ2A имел меньшую массу (сравнимую с массой удаленного фрагмента – 14 кДа) (рис. 6 а), равную таковой у полипротеина (L)P1. Указанные предшественники были обнаружены и с помощью радиоавтографии (рис. 6 б). Были выделены несколько сигналов, соответствующих вирусспецифичным белкам: № 1, 2, 3, 4. Анализ подвижности в полиакриламидном геле и данные литературы позволили предположить, что сигнал № 1 соответствует предшественнику неструктурных белков P3, № 2 – белку 3CD, № 3 – предшественнику VP4-VP2-VP3 (61 кДа); № 4 – полимеразе 3D (52 кДа). Можно обобщить, что сигналы 1–4 соответствуют либо белкам-предшественникам капсидных белков, либо неструктурным белкам.
Результаты иммуноблоттинга (рис. 6 а) показывают, что после 4 ч инфекции вирусами WT и Δ2A в клетках HeLa синтезировалось и накопилось больше вирусных белков, чем в случае инфекции Zfmut и ZfmutΔ2A. Количественный анализ результатов иммуноблоттинга также свидетельствует о том, что синтез всех детектируемых белков при введении мутации в L существенно ингибируется (рис. 7). Наблюдаемый факт, вероятно, связан с тем, что полная блокировка трансляции в клетке, инфицируемой мутантами по L, произошло на 1 ч раньше (к 4 ч вместо 5 ч) (рис. 4, 5 а), поэтому трансляция генома вирусов, мутантных по белку L, шла на 1 ч меньше. Можно заключить, что инактивация в составе генома белка L приводит к снижению эффективности трансляции вирусных белков.
Количественный анализ сигналов отдельных вирусных белков после окрашивания мышиной сывороткой лизатов зараженных клеток продемонстрировал особенности в эффективности накопления разных групп белков при частичной делеции 2А (рис. 7). Не было обнаружено отличий в накоплении полипротеина-предшественника (L)P1-2A у вируса WT и аналогичного ему (L)P1-Δ2A у мутантного вируса Δ2A, а также в эффективности накопления белкового продукта № 4 у этих вирусов (рис. 7 а). Мы наблюдали умеренное снижение накопления белкового продукта № 3 мутантного вируса Δ2A по сравнению с WT (рис. 7 а). Предшественник капсидных белков VP0, расщепляющийся на VP2 и VP4, накапливается в клетках HeLa, зараженных Δ2A, также с немного сниженной эффективностью, чем при инфекции WT (рис. 7 б). Накопление отдельных капсидных белков VP1, VP2 и VP3 вируса Δ2A существенно ингибировано (на 50–80%, рис. 6 а, 7 б). Сигналы вирусных белков на радиоавтографе также показывают существенное угнетение образования капсидных белков VP1, VP3 и предшественника VP0 в клетках, зараженных Δ2A, по сравнению с инфекцией WT (рис. 6 б). Стоит заметить, что при сравнении эффективности накопления вирусных белков в клетках HeLa, зараженных Zfmut и ZfmutΔ2A, также заметно уменьшение сигнала белков капсида у «двойного» мутанта при равенстве синтеза остальных белков (рис. 6, 7). Таким образом, синтез полипротеинов-предшественников капсидных белков и неструктурных белков при частичной делеции 2А не изменяется либо снижается умеренно. Образование отдельных белков, формирующих зрелый капсид вируса при делеции 2А, существенно ингибировано. Полученные данные указывают на то, что при частичной делеции белка 2А нарушается процессинг капсидных белков.
Обсуждение
«Секьюрити»-белки L и 2А представителей семейства Picornaviridae имеют множество функций в зараженной клетке [15]. Одной из них является участие в подавлении кэп-зависимой клеточной трансляции, необходимой для эффективного синтеза вирусных белков и продуктивного течения вирусной инфекции. Этот функциональный аспект хорошо изучен для белков, имеющих ферментативную активность внутри клетки, таких как Lpro вируса ящура или 2Аpro энтеровирусов. Проявляя протеолитическую специфическую активность в зараженных клетках, они расщепляют трансляционные факторы, необходимые для кэп-зависимой трансляции, которые не нужны для IRES-зависимой: eIF4G и PABP [17, 18, 40]. Белки L и 2А представителей рода Cardiovirus, к которому относится серотип EMCV-1, не имеют известной ферментативной активности, eIF4G и PABP не расщепляются при инфекции, однако ингибирование трансляции происходит [20]. Однако белки 2А и L EMCV являются главными кандидатами на роль подавления клеточного синтеза [21, 22, 34]. Как правило, для доказательства действия того или иного белка авторы предыдущих исследований инактивировали изучаемый белок путем введения мутаций или делеций участков белков. В своей работе мы придерживались аналогичной стратегии. Функциональную инактивацию белка L проводили путем внесения аминокислотных замен в Zn-связывающий домен (Cys19/Ala и Cys22/Ala), который ответственен за многие известные функции L: подавление развития апоптотической программы, ингибирование синтеза интерферонов, нарушение ядерно-цитоплазматического транспорта [25, 26, 41, 42]. Введение указанных мутаций используется в большинстве исследований функциональности белка L. В работе, посвященной роли L EMCV в подавлении клеточной трансляции, был использован мутантный вирус с делецией (с 12 по 52 а.о.) [22]. В нашей предыдущей работе мы показали, что инфекция мутантными вирусами с точечными заменами (Zfmut) и частичной делецией ΔL приводит к одинаковому изменению синтеза белков в клетке [38]. Таким образом, применение обеих мутантных моделей для исследования трансляционных особенностей зараженной клетки можно считать допустимым. Следует заметить, что использование модели с точечными заменами в белке L (Zfmut) является более предпочтительным, так как жизнеспособность такого мутанта, показанная нами ранее [38], снижена умеренно (в 5–10 раз, рис. 3 а) относительно WT. В то время как жизнеспособность мутантов с частичной делецией L снижена более чем в 100 раз [22, 38]. Поскольку основой работы по изучению трансляционных процессов в зараженных клетках является сравнение свойств мутантных вирусов с вирусом дикого типа, наиболее предпочтительной была модель, в которой мутантные вирусы способны размножаться с эффективностью, как можно более близкой к исходному WT. Это позволяет минимизировать возможные эффекты, которые возникают при чрезмерном угнетении репликации пикорнавирусов [43].
Нарушение функциональной активности белка 2А в нашей работе мы осуществляли путем удаления участка с 11 по 125 а.о. В нашей конструкции в белке сохраняются N- и С-концевые фрагменты, последовательность которых необходима для процессинга. С-концевой фрагмент (c 126 по 143 а.о.) содержит мотив NPG(P), где между остатками глицина (G) и пролина (P) осуществляется контрансляционный процессинг между молекулами (L)-P1-2A и 2B [28]. N-концевой фрагмент содержит часть сайта гидролиза вирусной протеазы 3Сpro (E-S), посредством которого от предшественников капсидных белков отщепляется белок 2А [39]. Полученные мутантные вирусы Δ2A и ZfmutΔ2A не имеют в составе белка 2А функционального участка с 94 по 102 а.о., играющего ключевую роль в осуществлении −1 рибосомального сдвига, контролирующего эффективность трансляции вирусных белков [31, 33]. Более того, этот фрагмент, по-видимому, направляет белок 2А в ядро [29] и участвует в связывании белка 2А с субъединицами рибосом [33]. Δ2A и ZfmutΔ2A также лишены потенциального сайта в составе белка 2А, ответственного за взаимодействие с белком L [44]. Таким образом можно с уверенностью предположить, что оставшаяся в мутантных вирусах часть белка 2А лишена известной функциональной активности. Подобный подход для изучения свойств был использован ранее, например, в исследовании роли белка 2А в подавлении клеточной трансляции [34], где были конструированы два мутантных вируса с частичным удалением белка 2А (с 39 по 97 а.о. и с 5 по 125 а.о.). В исследовании Ю.В. Свиткина и соавт. мутантные вирусы обладали существенно сниженной жизнеспособностью (в 100–1000 раз по сравнению с WT) в исследуемой культуре клеток [34]. Полученные нами мутанты были адаптированы путем проведения пассажей и клонированы (рис. 1). Судя по данным анализа последовательности геномов, адаптация затронула позицию 99 белка VP2 (замена остатка треонина на аланин, рис. 2).

Рис. 3. Накопление инфекционных вирусных частиц (a) и копий вирусной геномной РНК (б) в ходе цикла репродукции вирусов WT, Zfmut, Δ2A и Zfmut&Δ2A в клетках HeLa. Количество бляшкообразующих единиц (БОЕ) определяли методом титрования в клетках BHK-21. Количество копий вирусной РНК определяли методом ПЦР в режиме реального времени.
Fig. 3. Accumulation of infectious viral particles (a) and copies of viral genomic RNA (b) during the reproduction cycle of WT, Zfmut, Δ2A and Zfmut&Δ2A viruses in HeLa cells. The number of PFU was determined by plaque titration in BHK-21 cells. The number of copies of viral RNA was determined by real-time PCR.
Благодаря этому полученные мутантные вирусы по белку 2А: Δ2A и ZfmutΔ2A имеют умеренно сниженную репликативную активность: в ~5 и ~20 раз соответственно (рис. 3 а). Для исследования функций отдельных вирусных белков адаптированные вирусные мутанты, имеющие схожие репликативные свойства с исходным вирусом, более предпочтительны, чем практически не размножающиеся мутанты [34]: концентрация остальных вирусных белков будет существенно снижена, что может влиять на течение инфекции [43].
Наши эксперименты по анализу течения синтеза белков в зараженных клетках HeLa впервые выявили две стадии ингибирования трансляционных процессов при инфекции EMCV. Причем обе из них зависят от наличия функционального белка L. На первом этапе, спустя 3–4 ч после начала инфекции, наступает угнетение синтеза клеточных белков (на ~40%, рис. 4, 5, WT, Δ2A), при этом не наблюдается повышение уровня фосфорилирования eIF2α. При наличии слабого сигнала фосфорилирования в незараженных клетках (Mock) через 3 ч после начала инфекции WT и Δ2A сигнал eIF2α-P отсутствует, что говорит о вирус-опосредованном подавлении этого процесса (рис. 4, 5). При инфицировании вирусами, мутантными по белку L, не наблюдается как специфичного подавления клеточной трансляции, так и угнетения уровня eIF2α-P ниже базового, соответствующего незараженным клеткам (рис. 4, 5, Zfmut, ZfmutΔ2A). На более поздний срок инфекции приходится вторая неспецифическая стадия изменения синтеза белков. Она заключается в полном ингибировании трансляции, как клеточной, так и вирусной, сопровождающейся повышением уровня фосфорилирования eIF2α (рис. 4, 5). В отсутствие активной формы L вторая стадия наступает раньше. Результаты эксперимента свидетельствуют о том, что белок L принимает непосредственное участие в специфическом подавлении синтеза клеточных белков на начальном этапе инфекции, а также влияет на уровень фосфорилирования eIF2α. Первый тезис согласуется с предыдущим исследованием о роли белка L в подавлении клеточной трансляции [22]. Второго этапа подавления трансляции авторы не обнаружили, вероятно, не дождавшись поздней стадии инфекции, которая, по-видимому, наступает существенно позднее, чем в случае клеток HeLa [22]. Вторая, неспецифическая, стадия ингибирования синтеза белков в зараженной клетке, по-видимому, осуществляется за счет активации клеточных киназ, фосфорилирующих eIF2α. В составе вирусного генома киназ не обнаружено. Данный процесс является частью клеточных программ контроля инициации клеточной трансляции в ответ на различные сигналы и осуществляется киназами PKR, PERK, HRI и GCN2 [8].
Таким образом, один или несколько указанных ферментов могут участвовать в процессе блокировки трансляционных процессов в клетках HeLa, зараженных EMCV, причем белок L, по имеющимся данным, способен влиять на работу этих киназ. В присутствии L работа киназ на время ингибируется, откладывая образование eIF2α-P на 1 ч, а в отсутствие «работоспособного» белка L подобного ингибирования не обнаруживается (рис. 4, 5). Наиболее вероятно, что наблюдаемое в ходе инфекции EMCV фосфорилирование eIF2α, которое происходит раньше в отсутствие белка L, вызвано активностью киназы PKR. Именно этот фермент участвует в противовирусной защите клетки, активируясь в ходе интерферонового ответа [45], а также после появления в клетке двухцепочечной РНК, в том числе вирусной [46]. Показано, что в ходе инфекции EMCV нарушается активация киназы PKR [47], а белок L способен подавлять выработку интерферонов и моделировать каскады MAPK, участвующие в регуляции PKR [8, 27, 48]. Киназа HRI активируется и фосфорилирует eIF2α при снижении концентрации гема, тем самым регулируя синтез белков в ретикулоцитах [49]. Об активации этой киназы в ходе пикорнавирусной инфекции ничего не известно. Киназа GCN2 активируется в ответ на возникновение дефицита аминокислот в цитоплазме клеток. Показано, что она может быть активирована и в ответ на развитие вирусных инфекций, благодаря взаимодействию со специфичными РНК-структурами в составе генома, например вируса Синдбис и вируса везикулярного стоматита [50, 51]. Потенциально GCN2 может участвовать в ингибировании трансляции при инфекции EMCV, однако прежде указаний на это не было. Киназа PERK является одним из сенсорных белков, активирующихся при развитии ER-стресса (стресс эндоплазматического ретикулума) в ответ на накопление неправильно свернутых белков [52]. Развитие вирусной инфекции может способствовать активации ER-стресса. Нами ранее показано, что это происходит при заражении клеток энтеровирусами [53], следовательно, нельзя исключать роль PERK в наблюдаемом фосфорилировании eIF2α (рис. 4, 5). Однако данные об активации PERK при инфекции представителями рода Cardiovirus ранее отсутствовали. Таким образом, в настоящей работе был выявлен еще один эффект, вызываемый белком L, который связан с изменением течения реакций фосфорилирования клеточных белков, помимо участия в модификации белков ядерной поры [41].
Обнаруженная активность белка L свидетельствует о его важной роли в обеспечении эффективной трансляции вирусных белков. Мы показали, что нарушение функциональности белка L путем внесения двух аминокислотных замен в домене Zn-палец приводит к существенному сокращению эффективности накопления всех вирусных белков (рис. 7). По всей видимости, это происходит из-за активации киназных каскадов, приводящих к фосфорилированию eIF2α и ингибированию вирусной трансляции. Наши данные согласуются с ранее описанным эффектом, обнаруженным при использовании другого мутантного EMCV по белку L [24].
Результаты наших экспериментов однозначно указывают на отсутствие какой-либо роли белка 2А не только в подавлении клеточной трансляции, но и какой-либо модуляции трансляционных процессов в клетках HeLa, зараженных EMCV. Частичное удаление белка 2А, которое затронуло важнейшие функциональные участки белка, включая участок взаимодействия с субъединицей рибосом [33], не привело к каким-либо изменениям в течении трансляции клеточных белков при инфекции (рис. 4, 5).

Рис. 4. Радиоавтография синтезируемых белков в клетках HeLa, зараженных вирусами WT, Zfmut, Δ2A и Zfmut&Δ2A. Ингибиторы трансляции белков: Ars – арсенит натрия, концентрация 100 нМ, T-2 токсин – концентрация 50 нМ. Mock – незараженные клетки без воздействия ингибиторов. Сигналы, соответствующие вирусным белкам, отмечены стрелками.
Fig. 4. Autoradiography of synthesized proteins in HeLa cells infected with WT, Zfmut, Δ2A, and Zfmut&Δ2A viruses.
Protein translation inhibitors: Ars-sodium arsenite (100nM), T-2 toxin (50nM). Mock – uninfected cells without inhibitors. Signals corresponding to viral proteins are marked with arrows.

Рис. 5. Уровень включения аминокислот, меченных изотопом 14C, в синтезируемые белки в ходе инфекции клеток HeLa мутантными вирусами WT, Zfmut, Δ2A и Zfmut&Δ2A. Значение, равное 1, соответствует уровню включения аминокислот, меченных изотопом 14C, в белки незараженной культуры (а); уровень фосфорилирования фактора инициации трансляции eIFAα в клетках HeLa в ходе инфекции вирусами WT, Zfmut, Δ2A и Zfmut&Δ2A. Значение, равное 1, соответствует уровню фосфорилирования eIFAα в незараженных клетках (б). Значения получены при обработке результатов иммуноблоттинга. Обработка изображения проводилась с использованием программы ImageJ.
Fig. 5. The level of incorporation of 14C-labeled amino acids into synthesized proteins during infection of HeLa cells with WT, Zfmut, Δ2A and Zfmut&Δ2A viruses. A value of 1 corresponds to the level of incorporation of 14C-labeled amino acids into the proteins of an uninfected culture (a). Level of phosphorylation of translation initiation factor eIFAα in HeLa cells during infection with WT, Zfmut, Δ2A and Zfmut&Δ2A viruses. A value of 1 corresponds to the level of eIFAα phosphorylation in uninfected cells (b).
Values obtained by processing the results of immunoblotting. Image processing was carried out using the ImageJ program.
В соответствующих парах WT/Δ2A и Zfmut/Zfmut&Δ2A отсутствовала разница в характере изменения трансляционной активности с течением инфекции, а также в кинетике этого изменения. Наши результаты идут вразрез с выводами, сделанным по итогам нескольких исследований [21, 34].
В одном из них вывод о том, что 2А участвует в подавлении клеточной трансляции, был сделан на основе экспериментов, в которых клетки BHK-21 инфицировали мутантными EMCV, содержащими делеции в последовательности 2А [34]. Судя по данным, приведенным в исследовании, полученные мутантные вирусы обладали существенно сниженной способностью к размножению в клетках BHK-21. Разница в вирусном урожае по сравнению с WT составила 100–1000 раз. Сравнение вирусов, на несколько порядков различающихся по репродуктивной способности, требует дополнительных контролей, которые отсутствовали в описанной работе. К тому же по имеющимся в публикации данным сложно однозначно сделать вывод о клеточной трансляционной активности ввиду отсутствия доказательств об общем содержании белков (например, актина), необходимых для достоверного выравнивания образцов. Наша вирусная модель, в которой мутантные вирусы адаптированы к экспериментальной культуре клеток и показывают высокий уровень репродуктивной активности (рис. 3), является наиболее релевантной для исследований свойств вирусных белков.
Другое исследование, давшее начало теории о влиянии 2А EMCV на клеточную трансляцию, проведено на модели репликонов без использования жизнеспособных вирусов [21], что может лишь косвенно доказать участие того или иного белка в инфекционном процессе. Генетические конструкции, созданные в исследовании А.Г. Аминева и соавт., содержали ген люциферазы (кодирует 188 а.о.) под контролем эукариотического промотора и ген 2А EMCV (кодирует 143 а.о.) под контролем IRES. В исследовании было показано, что экспрессия люциферазы при использовании бицистронной конструкции, содержащей ген 2А, существенно снижается по сравнению с экспрессией люциферазы в конструкции без гена 2А. Описанные авторами результаты могут говорить о роли белка 2А в ингибировании трансляции люциферазы в рамках данной бицистронной конструкции. О глобальной роли 2А в клеточном синтезе белков при инфекции проведенные эксперименты свидетельствовать не могут.

Рис. 6. Изучение накопления белков исследуемых вирусов в ходе инфекции клеток HeLa с помощью специфической мышиной иммунной сыворотки (а) и радиоавтографии с использованием аминокислот, меченных изотопом 14С (б). Для панели а и б использовали одни и те же лизаты зараженных клеток. Образцы: 1 – WT EMCV-1, очищенный с помощью ультрацентрифугирования в градиенте хлорида цезия; 2 – WT из рабочего пула; 3–12 – образцы из клеток HeLa спустя 4 ч после заражения исследуемыми вирусами; Mock – незараженные клетки. Количество образцов 3–12 выровнено по содержанию β-актина.
Fig. 6. Study of the accumulation of proteins of the studied viruses during the infection of HeLa cells using specific mouse immune serum (a) and autoradiography using amino acids labeled with the 14C isotope (b).
For panels a and b, the same lysates of infected cells were used. Samples: 1 – WT EMCV-1 purified by cesium chloride gradient ultracentrifugation; 2 – WT from the working pool; 3–12 – samples from HeLa cells 4h after infection with the studied viruses; Mock – uninfected cells. The number of samples 3–12 are aligned according to the content of β-actin.
Необходимо отметить, что полученные нами результаты, свидетельствующие об отсутствие роли белка 2А в трансляционных процессах, согласуются с некоторыми экспериментальными данными исследователей, пытавшихся обнаружить функциональные участки 2А. В исследованиях, посвященных поиску домена 2А, который, вероятно, участвует в связывании eIF4E, было показано, что при введении множественных мутаций и делеций в аминокислотную последовательность 2А трансляционный паттерн зараженных клеток не меняется по сравнению с WT [29]. Введение мутаций в потенциальные активные сайты белка 2А, предсказанные с помощью in vitro подходов, не привело к каким-либо изменениям в динамике подавления клеточной трансляции [29], что полностью соотносится с результатами, показанными в нашем исследовании. Многочисленные свидетельства того, что белок 2А связывается с субъединицами рибосом [31, 35, 36], напрямую не означают, что он участвует в ингибировании синтеза белков. Полученные нами данные не могут опровергнуть этот факт. Использованные нами мутантные вирусы имеют делецию, которая включает обнаруженный ранее сайт взаимодействия с рибосомальной РНК (с 95 по 100 а.о.) [31, 33], поэтому мы можем предположить, что данное взаимодействие не влечет за собой изменение трансляционной активности. Вероятно, такое взаимодействие необходимо для функционирования 2А как индуктора −1 рибосомального сдвига [33].
Единственный эффект, который мы наблюдаем при частичном удалении белка 2А, заключается в нарушении накопления отдельных капсидных белков в ходе инфекции (рис. 7). Он, по всей видимости, связан с нарушением процессинга полипротеина-предшественника P1. Полученные результаты совпадают с данными предыдущих исследований, проводимых с мутантами EMCV по белку 2А [34]. Следует отметить, что этот эффект наблюдается только при делетирующих мутациях независимо от их размера, но отсутствует при точечных заменах аминокислот [29]. По-видимому, третичная структура белка 2А, которая нарушается при делециях, необходима для эффективного процесса отделения белков VP1, VP2 и VP3 от L-P1-2А.

Рис. 7. Относительный уровень сигнала вирусных белков, накопленных к 4 ч после начала инфекции клеток HeLa вирусами WT, Zfmut, Δ2A и Zfmut&Δ2A. а – уровень сигнала неструктурных белков и белков-предшественников капсидных белков; б – уровень сигнала капсидных белков. Данные получены в ходе обработки результатов иммуноблоттинга лизатов зараженных клеток с использованием специфической мышиной сыворотки против белков EMCV-1 (рис. 6 а). Уровень, равный 1, соответствует уровню сигнала каждого отдельного белка вируса WT. Величину сигнала отдельных белков нормировали на сигнал у вируса WT. Обработку изображения проводили с использованием программы ImageJ. * – p < 0,05, *** – p < 0,001.
Fig. 7. Relative signal level of viral proteins accumulated by 4 h.p.i. of HeLa cells with WT, Zfmut, Δ2A and Zfmut&Δ2A viruses.
a – signal level of non-structural proteins and precursor proteins of capsid proteins; b – signal level of capsid proteins. The data were obtained by processing the results of immunoblotting of lysates of infected cells using specific mouse serum against EMCV-1 proteins (fig. 6 a). a level of 1 corresponds to the signal level of each individual protein of the WT virus. The value of the signal of individual proteins was normalized to the signal of the WT virus. Image processing was carried out using the ImageJ program. * – p < 0.05, *** – p < 0.001.
Примечательно, что репликация генома Δ2A и Zfmut&Δ2A относительно WT и Zfmut соответственно остается неизменной (рис. 3 б), общий синтез неструктурных вирусных белков и белков-предшественников также остается на прежнем уровне (рис. 7). Отсюда мы можем предположить, что нарушение процессинга, вероятно, приводит к снижению урожайности вирусов, мутантных по белку 2А (рис. 3 а).
Суммируя все вышесказанное, мы показали, что среди «секьюрити»-белков EMCV-1 именно белок L играет ключевую роль в управлении трансляцией клеточных и вирусных белков. Белок 2А не участвует в изменении трансляционной активности в зараженных клетках.
1The Picornaviridae Pages. Available at: https://picornaviridae.com/
About the authors
Yury Yu. Ivin
Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute)
Author for correspondence.
Email: ivin_uu@chumakovs.su
ORCID iD: 0000-0003-0995-7944
researcher, Laboratory of biochemistry, Head of the Department of development and implementation innovative and pilot technology
Russian Federation, 108819, MoscowAnna A. Butusova
Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute)
Email: amadreaera@gmail.com
ORCID iD: 0009-0008-0361-9681
junior researcher, Laboratory of biochemistry
Russian Federation, 108819, MoscowEkaterina E. Gladneva
Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute)
Email: gladneva_ee@chumakovs.su
ORCID iD: 0009-0005-8067-6768
junior researcher, Laboratory of biochemistry
Russian Federation, 108819, MoscowGalina Ya. Kolomijtseva
A.N. Belozersky Research Institute of Physico-Chemical Biology MSU
Email: kolom@genebee.msu.ru
ORCID iD: 0009-0005-5130-2695
Cand. Sci. (Chem.), Assoc. Prof, Head of the Department of isotope analysis
Russian Federation, 119234, MoscowYusuf K. Khapchaev
Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute)
Email: hapchaev_uh@chumakovs.su
ORCID iD: 0000-0003-1613-5228
D. Sci. (Biol.), Head of the Department for the production of polio vaccines
Russian Federation, 108819, MoscowAydar A. Ishmukhametov
Federal State Autonomous Scientific Institution M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Drugs of the Russian Academy of Sciences (Polio Institute); Institute for Translational Medicine and Biotechnology, Sechenov First Moscow State Medical University
Email: ishmukhametov@chumakovs.su
ORCID iD: 0000-0001-6130-4145
D. Sci. (Med.), Professor, Academician of RAS, Head of the Department of organization and technology of production of immunobiological preparations
Russian Federation, 108819, Moscow; 117418, MoscowReferences
- Drappier M., Opperdoes F.R., Michiels T. Nonstructural protein L* species specificity supports a mouse origin for Vilyuisk human encephalitis virus. J. Virol. 2017; 91(14): e00573-17. https://doi.org/10.1128/jvi.00573-17
- Ugai S., Iwaya A., Taneichi H., Hirokawa C., Aizawa Y., Hatakeyama S., et al. Clinical characteristics of saffold virus infection in children. Pediatr. Infect. Dis. J. 2019; 38(8): 781–5. https://doi.org/10.1097/inf.0000000000002298
- Tan S.Z.K., Tan M.Z.Y., Prabakaran M. Saffold virus, an emerging human cardiovirus. Rev. Med. Virol. 2017; 27(1): e1908. https://doi.org/10.1002/rmv.1908
- Lipton H.L. Human Vilyuisk encephalitis. Rev. Med. Virol. 2008; 18(5): 347–52. https://doi.org/10.1002/rmv.585
- Carocci M., Bakkali-Kassimi L. The encephalomyocarditis virus. Virulence. 2012; 3(4): 351–67. https://doi.org/10.4161/viru.20573
- Merrick W.C., Pavitt G.D. Protein synthesis initiation in eukaryotic cells. Cold Spring Harb. Perspect. Biol. 2018; 10(12): a033092. https://doi.org/10.1101/cshperspect.a033092
- Jackson R.J., Hellen C.U.T., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010; 11(2): 113–27. https://doi.org/10.1038/nrm2838
- Donnelly N., Gorman A.M., Gupta S., Samali A. The eIF2α kinases: their structures and functions. Cell. Mol. Life Sci. 2013; 70(19): 3493–511. https://doi.org/10.1007/s00018-012-1252-6
- Wek R.C. Role of eIF2α kinases in translational control and adaptation to cellular stress. Cold Spring Harb. Perspect. Biol. 2018; 10(7): a032870. https://doi.org/10.1101/cshperspect.a032870
- Imai S., Suzuki H., Fujiyoshi Y., Shimada I. Dynamically regulated two-site interaction of viral RNA to capture host translation initiation factor. Nat. Commun. 2023; 14(1): 4977. https://doi.org/10.1038/s41467-023-40582-6
- Sorokin I.I., Vassilenko K.S., Terenin I.M., Kalinina N.O., Agol V.I., Dmitriev S.E. Non-canonical translation initiation mechanisms employed by eukaryotic viral MRNAs. Biochemistry. (Mosc.). 2021; 86(9): 1060–94. https://doi.org/10.1134/S0006297921090042
- Francisco-Velilla R., Embarc-Buh A., Abellan S., Martinez-Salas E. Picornavirus translation strategies. FEBS Open Bio. 2022; 12(6): 1125–41. https://doi.org/10.1002/2211-5463.13400
- Chamond N., Deforges J., Ulryck N., Sargueil B. 40S recruitment in the absence of EIF4G/4A by EMCV IRES refines the model for translation initiation on the archetype of type II IRESs. Nucleic Acids Res. 2014; 42(16): 10373–84. https://doi.org/10.1093/nar/gku720
- Welnowska E., Sanz M.A., Redondo N., Carrasco L. Translation of viral MRNA without active EIF2: The case of picornaviruses. PLoS One. 2011; 6(7): e22230. https://doi.org/10.1371/journal.pone.0022230
- Agol V.I., Gmyl A.P. Viral security proteins: counteracting host defences. Nat. Rev. Microbiol. 2010; 8(12): 867–78.https://doi.org/10.1038/nrmicro2452
- Yang X., Hu Z., Fan S., Zhang Q., Zhong Y., Guo D., et al. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 2018; 14(2): e1006901. https://doi.org/10.1371/journal.ppat.1006901
- Chau D.H.W., Yuan J., Zhang H., Cheung P., Lim T., Liu Z., et al. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/P97/NAT1. Apoptosis. 2007; 12(3): 513–24. https://doi.org/10.1007/s10495-006-0013-0
- Azzinaro P.A., Medina G.N., Rai D., Ramirez-Medina E., Spinard E., Rodriguez-Calzada M., et al. Mutation of FMDV Lpro H138 residue drives viral attenuation in cell culture and in vivo in swine. Front. Vet. Sci. 2022; 9: 1028077. https://doi.org/10.3389/fvets.2022.1028077
- Turkki P., Laajala M., Flodström-Tullberg M., Marjomäki V. Human enterovirus group B viruses rely on vimentin dynamics for efficient processing of viral nonstructural proteins. J. Virol. 2020; 94(2): e01393-19. https://doi.org/10.1128/JVI.01393-19.
- Jen G., Detjen B.M., Thach R.E. Shutoff of HeLa cell protein synthesis by encephalomyocarditis virus and poliovirus: a comparative study. J. Virol. 1980; 35(1): 150–6. https://doi.org/10.1128/JVI.35.1.150-156.19
- Aminev A.G., Amineva S.P., Palmenberg A.C. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent MRNA translation. Virus Res. 2003; 95(1-2): 45–57. https://doi.org/10.1016/S0168-1702(03)00162-X
- Zoll J., Galama J.M., van Kuppeveld F.J., Melchers W.J. Mengovirus leader is involved in the inhibition of host cell protein synthesis. J. Virol. 1996; 70(8): 4948–52. https://doi.org/10.1128/jvi.70.8.4948-4952.1996
- Bacot-Davis V.R., Ciomperlik J.J., Basta H.A., Cornilescu C.C., Palmenberg A.C. Solution structures of mengovirus leader protein, its phosphorylated derivatives, and in complex with nuclear transport regulatory protein, RanGTPase. Proc. Natl Acad. Sci. USA. 2014; 111(44): 15792–7. https://doi.org/10.1073/pnas.1411098111
- Dvorak C.M., Hall D.J., Hill M., Riddle M., Pranter A., Dillman J., et al. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology. 2001; 290(2): 261–71. https://doi.org/10.1006/viro.2001.1193
- Romanova L.I., Lidsky P.V., Kolesnikova M.S., Fominykh K.V., Gmyl A.P., Sheval E.V., et al. Antiapoptotic activity of the cardiovirus leader protein, a viral “security” protein. J. Virol. 2009; 83(14): 7273–84. https://doi.org/10.1128/JVI.00467-09
- Bardina M.V., Lidsky P.V., Sheval E.V., Fominykh K.V., van Kuppeveld F.J.M., Polyakov V.Y., et al. Mengovirus-induced rearrangement of the nuclear pore complex: hijacking cellular phosphorylation machinery. J. Virol. 2009; 83(7): 3150–61. https://doi.org/10.1128/JVI.01456-08
- Hato S.V., Ricour C., Schulte B.M., Lanke K.H., de Bruijni M., Zoll J., et al. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 2007; 9(12): 2921–30. https://doi.org/10.1111/j.1462-5822.2007.01006.x
- Donnelly M.L.L., Gani D., Flint M., Monaghan S., Ryan M.D. The cleavage activities of aphthovirus and cardiovirus 2A proteins. J. Gen. Virol. 1997; 78(Pt. 1): 13–21. https://doi.org/10.1099/0022-1317-78-1-13
- Groppo R., Brown B.A., Palmenberg A.C. Mutational analysis of the EMCV 2A protein identifies a nuclear localization signal and an eIF4E binding site. Virology. 2011; 410(1): 257–67. https://doi.org/10.1016/j.virol.2010.11.002
- Napthine S., Ling R., Finch L.K., Jones J.D., Bell S., Brierley I., et al. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun. 2017; 8: 15582. https://doi.org/10.1038/ncomms15582
- Hill C.H., Pekarek L., Napthine S., Kibe A., Firth A.E., Graham S.C., et al. Structural and molecular basis for cardiovirus 2A protein as a viral gene expression switch. Nat. Commun. 2021; 12(1): 7166. https://doi.org/10.1038/s41467-021-27400-7
- Loughran G., Firth A.E., Atkins J.F. Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proc. Natl Acad. Sci. USA. 2011; 108(46): E1111–9. https://doi.org/10.1073/pnas.1102932108
- Caliskan N., Hill C.H. Insights from structural studies of the cardiovirus 2A protein. Biosci. Rep. 2022; 42(1): BSR20210406. https://doi.org/10.1042/BSR20210406
- Svitkin Y.V., Hahn H., Gingras A.C., Palmenberg A.C., Sonenberg N. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. J. Virol. 1998; 72(7): 5811–9. https://doi.org/10.1128/jvi.72.7.5811-5819.1998
- Medvedkina O.A., Scarlat I.V., Kalinina N.O., Agol V.I. Virus-specific proteins associated with ribosomes of Krebs-II cells infected with encephalomyocarditis virus. FEBS Lett. 1974; 39(1): 4–8. https://doi.org/10.1016/0014-5793(74)80003-7
- Groppo R., Palmenberg A.C. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J. Virol. 2007; 81(23): 13067–74. https://doi.org/10.1128/JVI.00185-07
- Duke G.M., Palmenberg A.C. Cloning and synthesis of infectious cardiovirus RNAs containing short, discrete poly(C) tracts. J. Virol. 1989; 63(4): 1822–6. https://doi.org/10.1128/jvi.63.4.1822-1826.1989
- Mikitas O.V., Ivin Y.Y., Golyshev S.A., Povarova N.V., Galkina S.I., Pletjushkina O.Y., et al. Suppression of injuries caused by a lytic RNA virus (mengovirus) and their uncoupling from viral reproduction by mutual cell/virus disarmament. J. Virol. 2012; 86(10): 5574–83. https://doi.org/10.1128/jvi.07214-11
- Parks G.D., Baker J.C., Palmenberg A.C. Proteolytic cleavage of encephalomyocarditis virus capsid region substrates by precursors to the 3C enzyme. J. Virol. 1989; 63(3): 1054–8. https://doi.org/10.1128/jvi.63.3.1054-1058.1989
- Joachims M., Van Breugel P.C., Lloyd R.E. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 1999; 73(1): 718–27. https://doi.org/10.1128/jvi.73.1.718-727.1999
- Lidsky P.V., Hato S., Bardina M.V., Aminev A.G., Palmenberg A.C., Sheval E.V., et al. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 2006; 80(6): 2705–17. https://doi.org/10.1128/JVI.80.6.2705-2717.2006
- Hato S.V., Sorgeloos F., Ricour C., Zoll J., Melchers W.J.G., Michiels T., et al. Differential IFN-alpha/beta production suppressing capacities of the leader proteins of mengovirus and foot-and-mouth disease virus. Cell. Microbiol. 2010; 12(3): 310–7. https://doi.org/10.1111/j.1462-5822.2009.01395.x
- Tolskaya E.A., Romanova L.I., Kolesnikova M.S., Ivannikova T.A., Smirnova E.A., Raikhlin N.T., et al. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 1995; 69(2): 1181–9. https://doi.org/10.1128/jvi.69.2.1181-1189.1995
- Petty R.V., Basta H.A., Bacot-Davis V.R., Brown B.A., Palmenberg A.C. Binding Interactions between the Encephalomyocarditis virus leader and protein 2A. J. Virol. 2014; 88(22): 13503–9. https://doi.org/10.1128/JVI.02148-14
- Meurs E., Chong K., Galabru J., Thomas N.S.B., Kerr I.M., Williams B.R.G., et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990; 62(2): 379–90. https://doi.org/10.1016/0092-8674(90)90374-N
- Cole J. Activation of PKR: an open and shut case? Trends Biochem. Sci. 2007; 32(2): 57–62. https://doi.org/10.1016/j.tibs.2006.12.003
- Ng C.S., Jogi M., Yoo J.S., Onomoto K., Koike S., Iwasaki T., et al. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 2013; 87(17): 9511–22. https://doi.org/10.1128/jvi.03248-12
- Porter F.W., Brown B., Palmenberg A.C. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J. Virol. 2010; 84(24): 12538–48. https://doi.org/10.1128/JVI.01484-09
- Han A.P. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001; 20(23): 6909–18. https://doi.org/10.1093/emboj/20.23.6909
- Berlanga J.J., Ventoso I., Harding H.P., Deng J., Ron D., Sonenberg N., et al. Antiviral effect of the mammalian translation initiation factor 2α kinase GCN2 against RNA viruses. EMBO J. 2006; 25(8): 1730–40. https://doi.org/10.1038/sj.emboj.7601073
- Krishnamoorthy J., Mounir Z., Raven J.F., Koromilas A.E. The eIF2α kinases inhibit vesicular stomatitis virus replication independently of eIF2α phosphorylation. Cell Cycle. 2008; 7(15): 2346–51. https://doi.org/10.4161/cc.6323
- Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999; 397(6716): 271–4. https://doi.org/10.1038/16729
- Shishova A., Dyugay I., Fominykh K., Baryshnikova V., Dereventsova A., Turchenko Y., et al. Enteroviruses manipulate the unfolded protein response through multifaceted deregulation of the Ire1-Xbp1 pathway. Viruses. 2022; 14(11): 2486. https://doi.org/10.3390/v14112486
Supplementary files

