



NGS amplification panel HCV-seq for sequencing hepatitis C virus RNA (Flaviviridae: Hepacivirus)
- Authors: Chanyshev M.D.1, Chernyshova A.S.1,2, Glushchenko A.G.1,2, Grishaeva A.A.1, Makashova V.V.1, Ponezheva Z.B.1, Khafizov K.F.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute of Epidemiology
- Moscow Institute of Physics and Technology
- Issue: Vol 70, No 4 (2025)
- Pages: 363-373
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16786
- DOI: https://doi.org/10.36233/0507-4088-331
- EDN: https://elibrary.ru/fbgobh
- ID: 16786

Cite item

Abstract
Introduction. Hepatitis C is a pressing global public health issue. The high variability of the hepatitis C virus (HCV) complicates its whole-genome sequencing; most studies sequence only specific regions of the genome. There is a need for a simple and reliable method for sequencing the whole genome of HCV.
Objective. Development and validation of NGS panel for whole-genome sequencing of HCV.
Materials and methods. This study presents NGS amplification panel for sequencing the genomes of HCV genotypes 1, 2, and 3. Depending on the genotype, a set of 79, 67, or 89 primers is used. These primers enable amplification of overlapping regions of the HCV genome.
Results. The panel was tested on 153 HCV RNA samples isolated from blood plasma specimens (93/6/54 samples of genotypes 1/2/3, respectively). Shannon entropy analysis showed that genetic heterogeneity within the E2 gene is significantly higher than in other parts of viral genome. The frequency of mutations associated with drug resistance was determined. Specifically, for genotype 1, the following mutation detection rates were observed in NS3: Y56F – 37.6%, V170I – 23.7%; in NS5a: R30Q – 8.6%, P58L/S/T – 6.5%, A92T – 4.3%; in NS5b: L159F – 45.2%, S556G/N – 33.3%.
Conclusion. The current study describes a method for whole-genome sequencing of HCV genotypes 1, 2, and 3. The HCV sequencing panel shows great potential for use in scientific research and epidemiological monitoring.
Keywords
Full Text
Introduction
Viral hepatitis C poses a serious problem for healthcare both in Russia and worldwide. According to estimates by the World Health Organization, in 2022, there were approximately 50 million people worldwide suffering from chronic hepatitis C (CHC) and 1.0 million new cases of infection. In 2020, 24.5 thousand patients with a diagnosis of CHC were registered in Russia, and the incidence rate was 16.7 per 100 thousand people [1]. As of 2024, approximately 2.9% of the population of the Russian Federation is presumed to be infected with the hepatitis C virus (HCV). On average, 70% of those infected develop a chronic form of the disease, which can lead to cirrhosis of the liver (CL) and hepatocellular carcinoma (HCC). According to the results of the analysis of incidence, prevalence, and mortality of HCV infection, 16,427 deaths due to complications of CHC were registered in Russia in 2015 [2].
The pathogen of the disease is an RNA-containing virus from the Flaviviridae family (Flaviviridae: Hepacivirus: Hepacivirus hominis). The HCV genome is represented by single-stranded RNA and demonstrates high variability. There are 7 main genotypes of HCV known, with nucleotide sequence differences between genotypes ranging from 30% to 35%. The frequency of genotype occurrence varies across different regions of the world: genotype 1 predominates in Europe, North and South America, genotype 3 in India and several Southeast Asian countries. In the territory of the Russian Federation, almost all typed samples belong to genotype 1 (approximately 70% of cases), 2 (~ 5%) and 3 (~ 20%) [3, 4].
Numerous studies show that various genetic variants of HCV and mutations in different regions of the viral genome may be associated with the clinical features of the disease course and its outcomes, as well as resistance to therapy. For example, it was noted that genotype 1b was characterized by a high risk of cirrhosis and hepatocellular carcinoma [5]. Amino acid substitutions responsible for HCV drug resistance are predominantly located in genes encoding viral non-structural proteins such as NS3, NS5a and NS5b [6]. HCV-resistant variants are found in the majority of patients who have not achieved a sustained virological response due to the selection and growth of HCV-resistant variants in the body of a specific carrier. These resistance-associated mutations depend on the class of direct-acting antiviral drugs (DAA) used and also vary depending on the genotype and subtype of HCV [7]. Other regions of the viral genome are also of interest. Deletion in NS2 may be associated with liver cirrhosis [8]. In a study by S. Fishman et al., mutations in core 36G/C, 209A, 271U/C, 309A/C, 435A/C, 481A and 546A/C were associated with a high risk of HCC [9].
In most studies involving HCV RNA sequencing, individual regions of the viral genome were determined [9–11]. To date, a small number of studies have been published that have determined the complete genome of HCV, which may be related to the high genetic variability of this virus. For example, one study from 2019 included only 2 samples and represented the first complete readings of HCV in Latin America. During the study, the nested polymerase chain reaction (PCR) method was used, followed by Sanger sequencing of the amplified regions [12]. A similar approach for amplification was used in the study to compare the sensitivity of whole-genome sequencing of HCV genomes on the PacBio and Illumina platforms for detecting low-frequency mutations [13]. A similar sample preparation and sequencing on the Illumina MiSeq were performed for 88 samples with the aim of obtaining data on the distribution of genotypes in the London sample [14].
In Russia, HCV sequencing is primarily conducted to determine the viral genotype, with only individual enzyme genes being analyzed using the Sanger sequencing method. For example, V.O. Kotova et al. determined the nucleotide sequence of the NS5b gene in 60 HCV samples [15]. In a study by D.E. Reinhardt et al., the NS3, NS5a and NS5b genes were sequenced in 31 samples [16]. V.A. Isakov et al. analyzed the occurrence of substitutions in the NS3, NS5A and NS5B genes associated with drug resistance in samples from the Russian population, with sequencing based on Illumina MiSeq performed in the USA [17]. It is estimated that, to date, there is an extremely limited number of studies worldwide in which the authors have sequenced the whole genome of HCV using NGS. On the territory of the Russian Federation, no such publications were found.
The aim of the study is to develop an NGS amplification panel for whole-genome sequencing of HCV genotypes 1, 2 and 3 and to conduct its testing.
Materials and methods
Samples
For the study, 153 blood plasma samples were selected from participants of a clinical trial (protocol No. 133 dated March 2, 2023, of the Ethics Committee of the Central Research Institute of Epidemiology of Rospotrebnadzor) with viral hepatitis C. All study participants provided voluntary informed consent for the use of the samples. HCV RNA was extracted from plasma using the RIBO-prep kit (AmpliSens, Russia). The presence of HCV RNA and genotype were determined using the HCV-Monitor-FL and AmpliSens HCV-1/2/3-FL reagent kits (AmpliSens, Russia). Of these, 93 samples were identified as genotype 1, 6 as genotype 2, and 54 as genotype 3.
Primer selection
The performed bioinformatics analysis showed the impossibility of selecting universal primer sequences for whole-genome amplification of HCV simultaneously for all three main genotypes of the virus circulating in Russia. At the same time, it is possible to select three sets of primers that would be applicable to each of the three genotypes individually. For each genotype, primers were selected in a semi-automatic mode, as previously described for the development of an amplification panel for HCV genome sequencing [18]. As a result, 79/67/89 primers were selected for genotypes 1/2/3, respectively. Three control samples corresponding to genotypes 1, 2, and 3 of HCV (HCV1_01, HCV2_01, HCV3_01) were sequenced several times in succession, resulting in the selection of optimal PCR conditions and the concentration of each primer in the reaction mixture. The primer sequences are provided in Table S1, presented in the Appendix.
HCV sequencing
The REVERTA-L kit (AmpliSens, Russia) was used for reverse transcription. Three sets of primers were used for the amplification of the HCV genome, each corresponding to genotype 1, 2 or 3. The genotypes of the samples were determined in advance. For each sample, 5 separate multiplex PCR were performed, each reaction contained 10 µL of complementary DNA (cDNA), 10 µL of PCR-mix-2-blue PCR mix (AmpliSens, Russia), 1.4 µL of 4.4 mM dNTPs (AmpliSens, Russia), primers from one of the 5 pools, and mQ water to a final volume of 25 µL. Table S1 in the Appendix contains information on the nucleotide sequence, concentration in the reaction mixture and pool number for each primer. The PCR stages had the following conditions: 1) 95 °C for 3 min; 2) 20 cycles: 95 °C for 30 s, 55 °C for 30 s, 72 °C for 40 s; 3) 72 °C for 3 min. Next, the products of 5 parallel PCR reactions were mixed in 5 µL aliquots and purified using AMPure XP Beads (Beckman Coulter, USA), with an AMPure XP Beads/PCR product ratio of 0.8: 1. Then, PCR indexing was performed, each reaction contained 5 µL of purified PCR products, 10 µL of PCR-mix-2-blue (AmpliSens, Russia), 1.4 µL of 4.4 mM dNTPs (AmpliSens, Russia), standard Nextera index adapters (concentration in the reaction mixture amounted to 200 nM), and mQ water to a final volume of 25 µL. The PCR stages had the following conditions: 1) 95 °C for 3 min; 2) 25 cycles: 95 °C for 20 s, 55 °C for 30 s, 72 °C for 40 s; 3) 72 °C for 3 min. The indexed PCR products were mixed and subsequently purified using AMPure XP Beads 0.8/1. The Qubit dsDNA HS Assay Kit (Invitrogen, USA) was used to measure the concentration of the obtained library. Sequencing was performed using the MiSeq v. 3 (600 cycles) kit on the Illumina MiSeq.
Bioinformatics
To assess the quality of the obtained reads, the FastQC program was used. Adapter sequences were removed using the Trimmomatic program. To remove primer sequences, the Cutadapt program was used. Processed reads were mapped to the corresponding references (M58335.1/NC_009823.1/D17763.1 for genotypes 1/2/3) using the BWA-MEM program. Consensus sequences were obtained from bam files using iVar. To construct the phylogenetic tree, 42 reference HCV genomes from the euHCVdb database, representing genotypes 1 to 7, were used. Multiple sequence alignment was performed using MAFFT v. 7.505. The phylogenetic tree was constructed in MEGA v. 12.0.11 using the neighbor-joining algorithm and rooted using the midpoint method. The visualization was done using the iToL program.
Panel validation
For the validation of HCV-seq, three control samples of different genotypes were sequenced (HCV1_01, HCV2_01, HCV3_01) using an alternative approach. Reverse transcription was performed using the SuperScript IV Reverse Transcriptase kit (ThermoFisher, USA) according to the manufacturer’s recommendations. For the amplification of HCV RNA, a separate set of primers was used for each sample (listed in Table S2 in the Appendix). The PCR mixture contained 10 µL of template cDNA, 12.5 µL of Q5 High-Fidelity DNA Polymerase MasterMix 2x (NEB, USA), primers at a final concentration of 200 nM, and mQ water to a final volume of 25 µL. The PCR stages consisted of the following conditions: 1) 98 °C for 1 min; 2) 40 cycles: 98 °C for 10 s, 60 °C for 30 s, 72 °C for 2 min; 3) 72 °C for 3 min. The PCR products were checked using electrophoresis in a 1% agarose gel. The purification of PCR products was carried out using AMPure XP Beads at a ratio of 0.5 : 1. The concentration of the PCR products was determined using the dsDNA HS Assay Kit (Invitrogen, USA). Next, for each sample, the PCR products were diluted to 1 ng/μL and mixed in equimolar amounts. Libraries for sequencing were prepared using the Nextera XT DNA kit (Illumina) according to the manufacturer’s recommendations. Sequencing on the Illumina platform and genome assembly were performed as described above. Furthermore, for the sample HCV2_01 (genotype 2), the genomic sequence was also confirmed by Sanger sequencing. The sequencing results of the samples using HCV-seq, Nextera, and Sanger were almost completely identical. The raw NGS read files using HCV-seq and Nextera for all three samples, as well as the Sanger sequencing data for sample HCV2_01, can be downloaded at https://doi.org/10.5281/zenodo.15846624.
Results
As part of this work, 153 HCV genomes were sequenced and analyzed. The obtained genomes are presented at https://doi.org/10.5281/zenodo.15846624 in the form of HCV_SAMPLES.fasta. The average coverage of the viral genome was 96.2% of the area defined by the primers (Fig. 1). The read depth of amplicons for the three genotypes is presented in Fig. 2. Incomplete coverage of the samples may be associated with the high variability of HCV and the presence of mutations at the primer binding sites, as well as with the insufficient quantity and quality of HCV RNA.

Fig. 1. Coverage of 153 samples.
The genome region was considered as read when coverage was at least 10 reads. The coverage percentage was calculated for the region flanked by primers. Samples are sorted by coverage percentage and genotype.
Рис. 1. Покрытие 153 образцов.
Область генома считали прочитанной при глубине не менее 10 прочтений. Процент покрытия считали от области, ограниченной праймерами. Образцы отсортированы по проценту покрытия и генотипу.

Fig. 2. Log10 of the reads per amplicon normalized by total reads per sample.
a – genotype 1, b – genotype 2, c – genotype 3. The X-axis shows the amplicons, the Y-axis shows the samples. Unread (yellow) regions, singly distributed across genomes and samples, are associated with multiple mutations at the primer annealing sites in a given samples.
Рис. 2. Log10 глубины прочтения для каждого ампликона, нормированной на общее количество прочтений на образец.
а – генотип 1, б – генотип 2, в – генотип 3. По оси X отмечены ампликоны, по оси Y – образцы. Непрочитанные (желтые) участки единично распределены по геномам и образцам и связаны с множественными мутациями на местах отжига праймера в конкретном образце.
Phylogenetic analysis (Fig. 3) confirmed the precise separation of all studied samples by genotypes: genotype 1 samples (red), genotype 2 (blue), and genotype 3 (green) formed stable, well-defined clusters. All sequenced isolates reliably clustered with the reference sequences of their genotypes, confirming the accuracy of the preliminary genotyping. At the same time, clear clustering by subtypes was observed: genotype 1 with subtype 1b, genotype 2 with subtypes 2a and 2c, and genotype 3 with subtype 3a.

Fig. 3. Phylogenetic tree of sequenced HCV samples.
The genotype colors correspond to [3]. Reference sequences are labeled in black, while sequences obtained in this study are highlighted in blue.
Рис. 3. Филогенетическое дерево секвенированных образцов ВГC.
Цвета генотипов соответствуют [3]. Референсные последовательности отмечены черным цветом, а секвенированные в данной работе – синим.
To assess nucleotide variability, a Shannon entropy analysis was conducted, reflecting the degree of uncertainty in the distribution of nucleotides across positions. Fig. 4 shows the average Shannon entropy values calculated for each genotype based on non-overlapping blocks of 40 nucleotides. Statistical analysis showed that the mean Shannon entropy within the E2 gene is significantly higher than in the rest of the genome in all three analyzed HCV genotypes (p < 0.0001 by the Mann–Whitney test). This confirms the observed peak of variability in this region, especially in the 5’ region of the E2 gene. Increased entropy in the E1 regions and especially E2 reflects the virus’s adaptation to immune pressure. It is known that the 5’-end region of the E2 gene encodes the N-terminal hypervariable region HVR1, which is associated with evasion from neutralizing antibodies and interaction with host cell receptors [19].

Fig. 4. Average Shannon entropy for non-overlapping 40-nucleotide regions of the genome.
a – Shannon entropy profile for genotype 1, b – Shannon entropy profile for genotype 2, c – Shannon entropy profile for genotype 3. D – Schematic representation of the HCV genome with genome region boundaries indicated. Dashed lines indicate the boundaries of the E1 and E2 regions.
Рис. 4. Среднее значение энтропии Шеннона для неперекрывающихся участков генома длиной 40 нуклеотидов.
a – профиль энтропии Шеннона для генотипа 1, б – профиль энтропии Шеннона для генотипа 2, в – профиль энтропии Шеннона для генотипа 3, D – схематическое представление генома ВГС с обозначением границ генов. Пунктирные линии обозначают границы генов E1 и E2.
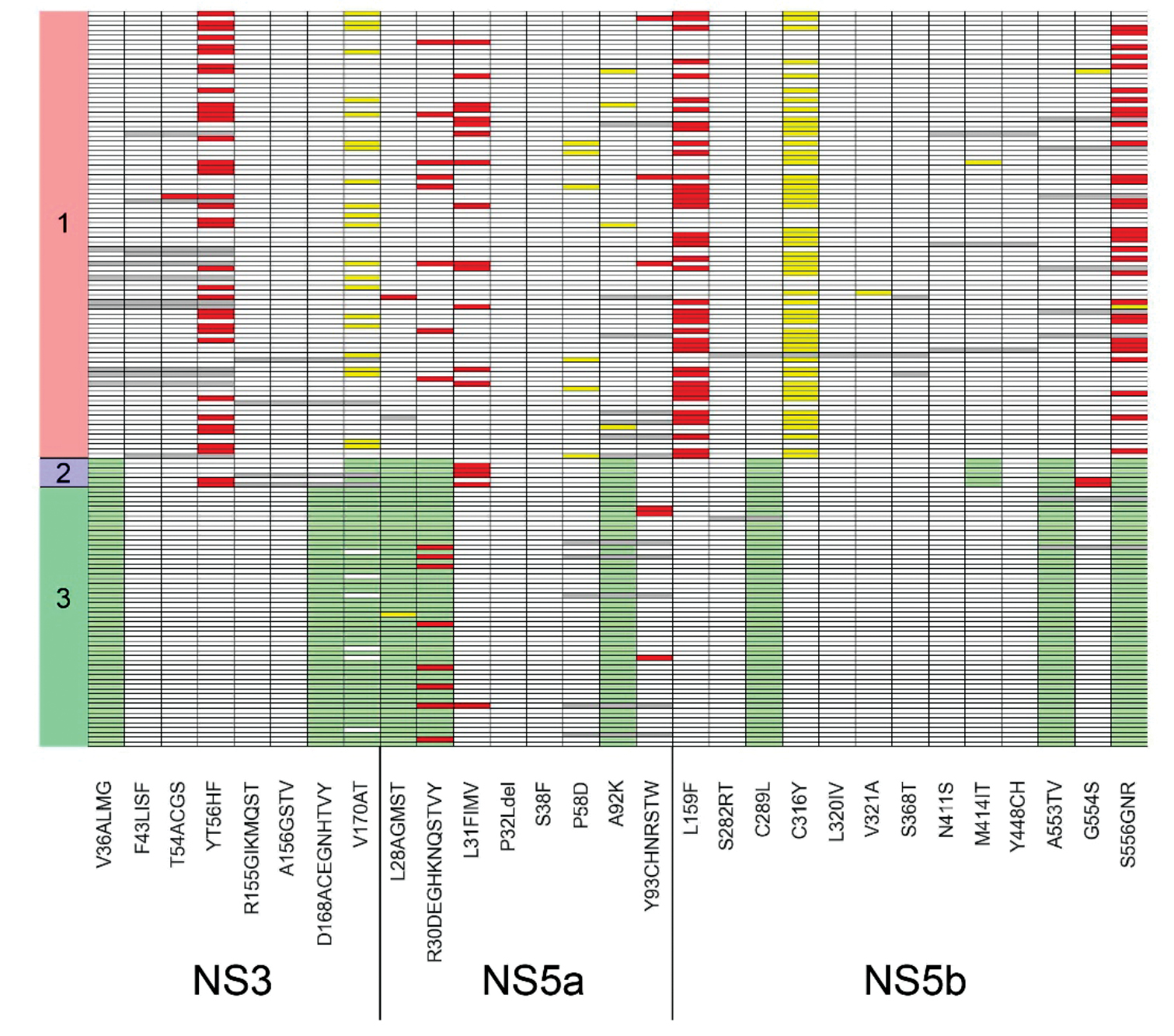
Next, an assessment of the incidence of HCV mutations associated with drug resistance was conducted. The correspondence of mutations and drugs was carried out according to the study1. The detected mutations are presented in the form of a diagram in Fig. 5. For genotype 1, the following mutation detection rates were noted in NS3: Y56F – 37.6%, V170I – 23.7%; in NS5a: R30Q – 8.6%, P58L/S/T – 6.5%, A92T – 4.3%; in NS5b: L159F – 45.2%, S556G/N – 33.3%. For genotype 2 samples, mutations characteristic of this genotype were noted – V36L and V170I in NS3, L28C/F, R30K and A92C/S in NS5a, and C289M, M414Q, A553V, and S556G in NS5b. Furthermore, substitutions Y56F in NS3, L31M in NS5a, and G554S in NS5b were also observed. For genotype 3 samples, characteristic mutations for this genotype were also identified: V36L, D168Q, and V170I in NS3, L28M, R30A and A92E in NS5a, C289F, A553V and S556G in NS5b. For genotype 3, the following mutation detection rates in NS5a were noted: R30K/S/T – 14.8%, L31I – 1.9%, and Y93H – 5.6%. No resistance mutations to the NS3/4A inhibitor group drugs glecaprevir and paritaprevir were detected in any of the samples. At the same time, HCV genotype 1 samples contained possible resistance mutations to NS5A inhibitor group drugs (daclatasvir, elbasvir, ledipasvir, velpatasvir), which were found in 95.7% of the samples, while for the NS5B inhibitor group (sofosbuvir and dasabuvir), the mutation detection rate was 45.1%. Samples of HCV genotype 2 contained possible resistance mutations to NS5A inhibitor group drugs in 66.7% of samples, while no resistance mutations were detected for NS5B inhibitor group drugs. HCV genotype 3 samples contained possible resistance mutations to NS5A inhibitor group drugs in 15.1% of samples, while resistance mutations to NS5B inhibitor group drugs were not detected.

Fig. 5. The detection rates of mutations in the NS3, NS5a, and NS5b coding regions associated with resistance to therapy.
Gray fields represent undefined areas, white – wild type, green – mutations characteristic of the genotype, yellow – substitutions not described in the literature, and red – mutations described in the literature as mutations of resistance to antiviral therapy. The X-axis shows mutations and genome regions, the Y-axis shows samples and genotypes.
Рис. 5. Встречаемость мутаций в генах NS3, NS5a, NS5b, ассоциированных с резистентностью к терапии.
Серые поля представляют собой неопределенные области, белые – дикий тип, зеленые – мутации, характерные для данного генотипа, желтые – замены, не описанные в литературе, красные – мутации, описанные в литературе как мутации устойчивости к противовирусной терапии. По оси X отмечены мутации и гены, по оси Y – образцы и генотипы.
Discussion
This study presents a method of whole-genome sequencing of HCV based on PCR enrichment of relatively short amplicons. There are a few studies that include whole-genome sequencing of HCV. In a study by N. Spitz et al., nested PCR was used, and the length of the amplicons was approximately 4.5 kb. Next, the obtained amplicons were sequenced using Sanger sequencing with internal primers [12]. The studies by R. Bull et al. and D. Aisyah et al. involved the selection of primers to amplify the whole viral genome, and during the experiments, the amplicons were subsequently tagmented and sequenced on the Illumina platform [13, 14]. It should be noted that this approach involves obtaining long amplicons of 4–9 kb, which requires a sufficient amount of full-length viral genome copies in the sample, whereas many samples contain fragmented RNA. The use of short amplicons allows for the sequencing of a large portion of clinical samples. It is interesting to note that for the routine sequencing of millions of SARS-CoV-2 genomes worldwide, methods with amplification of relatively short viral genome regions, such as ARTIC and Midnight, were used [20, 21]. The method presented in this study also has drawbacks. Independent amplification of short segments of the viral genome does not allow for the detection of extensive deletions in fractions of viral genomes, which were noted in a study by K. Yamauchi et al. [8].
The data obtained in the present study on the genotypic diversity of HCV in the sample of the Russian population generally agree with the few published results from other research groups. For example, in a study by N.N. Pimenov et al., conducted on 10,107 samples, the prevalence of HCV genotypes 1/2/3 in the Russian Federation was 53.6%, 7.6% and 35.4%, respectively [4]. The frequency of genotypes 1/2/3 in our relatively small sample of 153 specimens was 60.8/3.9/35.3%. D.E. Reinhardt et al. determined the nucleotide sequence of the NS3, NS5a and NS5b genes in 31 samples [16]. Overall, the types and detection rates of identified mutations in their study correlate with the results we obtained. For a number of mutations, the detection rate in a study by D.E. Reinhardt et al. was significantly higher. This may be related to the fact that the study material consisted of 31 blood plasma samples from patients with HCV who had a recurrence of the disease following DAA therapy, while a significant portion of our sample consisted of patients with newly diagnosed HCV. For example, the detection rate of NS5a Y93H was 3.9% in the present study and 41.9% in a study by D.E. Reingardt et al.
In the present study, the highest detection rate of resistance mutations was determined for drugs from the group of NS5A HCV protein inhibitors. This observation is consistent with the results of numerous published studies. Thus, it is known that NS5A protein inhibitors of HCV are most prone to the formation of resistance mutations. Studies show that mutations in the NS5A gene are found in a significant portion of patients (up to 30% in naive patients with genotype 1b) [22]. It should also be noted that there is significant cross-resistance among the currently available NS5A inhibitors [23]. Resistance mutations to NS5A and NS3 inhibitors often accumulate in patients without a sustained virological response after treatment with drugs from these groups. At the same time, NS3/4A and NS5B inhibitors are also used in therapy, but the frequency of resistance mutations to them is lower. For example, the NS5B inhibitor sofosbuvir has the highest barrier to resistance, and mutations to it are extremely rare. NS3/4A inhibitors can cause resistance mutations, but they are less stable compared to NS5A inhibitors [22]. Resistance mutations to NS5B inhibitors, according to various sources, are rarely detected (1% of cases) even after the use of an ineffective DAA regimen containing drugs from this group[2]. In the samples we studied, no mutations conferring resistance to NS3/4A inhibitor drugs were found, and mutations conferring resistance to NS5B inhibitor drugs were only observed among patients with HCV genotype 1. Thus, the integration of drug resistance testing into clinical practice can enhance the effectiveness of hepatitis C treatment. Resistance testing, especially to NS5A inhibitors, may be recommended for all patients who have not achieved a sustained virological response after using treatment regimens containing drugs from this group.
Conclusion
This study presents an amplification panel that enables whole-genome sequencing of HCV genotypes 1, 2 and 3. The panel is based on the amplification of relatively short regions of the viral genome, which allows for sequencing even highly fragmented samples, and has great potential for scientific and practical purposes. The widespread use of combined interferon-free regimens has achieved significant success in the treatment of HCV. Despite the fact that DAA provide a sustained virological response in more than 90% of cases, the issue of drug resistance is becoming increasingly relevant. The high replication rate of HCV and the low accuracy of its RNA polymerase lead to significant genetic variability within the viral population, which promotes the rapid emergence and selection of therapy-resistant variants. Monitoring the drug resistance of HCV is crucial for the successful treatment of patients with CHC. Determining mutations that cause resistance to DAA allows for the selection of the most effective therapy regimen and significantly increases the chances of complete recovery for the patient.
1 https://hcv.geno2pheno.org
2 AASLD. HCV Resistance Primer. HСV Guidance; 2022. Available at: https://hcvguidelines.org/evaluate/resistance
About the authors
Mikhail D. Chanyshev
Central Research Institute of Epidemiology
Author for correspondence.
Email: chanishq@gmail.com
ORCID iD: 0000-0002-6943-2915
PhD, Senior Researcher, Laboratory for Genomic Research
Russian Federation, 111123, MoscowAnastasia S. Chernyshova
Central Research Institute of Epidemiology; Moscow Institute of Physics and Technology
Email: chernyshova.a@cmd.su
ORCID iD: 0009-0000-2440-1309
Laboratory assistant, Laboratory for Genomic Research, Central Research Institute of Epidemiology
Russian Federation, 111123, Moscow; 141701, DolgoprudnyAlbina G. Glushchenko
Central Research Institute of Epidemiology; Moscow Institute of Physics and Technology
Email: albinagluschenko@gmail.com
ORCID iD: 0009-0002-8851-8703
Laboratory research assistant, Laboratory for Genomic Research, Central Research Institute of Epidemiology
Russian Federation, 111123, Moscow; 141701, DolgoprudnyAntonina A. Grishaeva
Central Research Institute of Epidemiology
Email: antoninagrishaeva@yandex.ru
ORCID iD: 0000-0002-1326-9274
PhD, Researcher of the Clinical Department of Infectious Pathology
Russian Federation, 111123, MoscowVera V. Makashova
Central Research Institute of Epidemiology
Email: veramakashova@yandex.ru
ORCID iD: 0000-0002-0982-3527
Dr. of Sci., Leading Researcher, Clinical Department of Infectious Diseases
Russian Federation, 111123, MoscowZhanna B. Ponezheva
Central Research Institute of Epidemiology
Email: doktorim@mail.ru
ORCID iD: 0000-0002-6539-4878
Dr. of Sci., Associate Professor, Head of the Clinical Department of Infectious Diseases
Russian Federation, 111123, MoscowKamil F. Khafizov
Central Research Institute of Epidemiology
Email: kkhafizov@gmail.com
ORCID iD: 0000-0001-5524-0296
PhD, Head of Laboratory for Genomic Research
Russian Federation, 111123, MoscowVasily G. Akimkin
Central Research Institute of Epidemiology
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
Academician of the Russian Academy of Sciences, Dr. of Sci., Professor, Director
Russian Federation, 111123, MoscowReferences
- Isakov V., Nikityuk D. Elimination of HCV in Russia: Barriers and perspective. Viruses. 2022; 14(4): 790. https://doi.org/10.3390/v14040790
- Pimenov N.N., Komarova S.V., Karandashova I.V., Tsapkova N.N., Volchkova E.V., Chulanov V.P. Hepatitis С and its outcomes in Russia: analysis of incidence, prevalence and mortality rates before the start of the programme of infection elimination. Infectious diseases: news, opinions, training. 2018; 16(3): 37–45. https://doi.org/10.20953/1729-9225-2018-3-37-45 https://elibrary.ru/ysjrqt (in Russian)
- Messina J.P., Humphreys I., Flaxman A., Brown A., Cooke G.S., Pybus O.G., et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015; 61(1): 77–87. https://doi.org/10.1002/hep.27259
- Pimenov N., Kostyushev D., Komarova S., Fomicheva A., Urtikov A., Belaia O., et al. Epidemiology and genotype distribution of hepatitis C virus in Russia. Pathogens. 2022; 11(12): 1482. https://doi.org/10.3390/pathogens11121482
- Raimondi S., Bruno S., Mondelli M.U., Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J. Hepatol. 2009; 50(6): 1142–54. https://doi.org/10.1016/j.jhep.2009.01.019
- Kalaghatgi P., Sikorski A.M., Knops E., Rupp D., Sierra S. Geno2pheno[HCV] – a Web-based interpretation system to support hepatitis C treatment decisions in the era of direct-acting antiviral agents. PLoS One. 2016; 11(5): e0155869. https://doi.org/10.1371/journal.pone.0155869
- Ahmed A., Felmlee D.J. Mechanisms of hepatitis C viral resistance to direct acting antivirals. Viruses. 2015; 7(12): 6716–29. https://doi.org/10.3390/v7122968
- Yamauchi K., Maekawa S., Osawa L., Komiyama Y., Nakakuki N., Takada H., et al. Single-molecule sequencing of the whole HCV genome revealed envelope deletions in decompensated cirrhosis associated with NS2 and NS5A mutations. J. Gastroenterol. 2024; 59(11): 1021–36. https://doi.org/10.1007/s00535-024-02146-3
- Fishman S.L., Factor S.H., Balestrieri C., Fan X., Dibisceglie A.M., Desai S.M., et al. Mutations in the hepatitis C virus core gene are associated with advanced liver disease and hepatocellular carcinoma. Clin. Cancer Res. 2009; 15(9): 3205–13. https://doi.org/10.1158/1078-0432.ccr-08-2418
- Costantino A., Spada E., Equestre M., Bruni R., Tritarelli E., Coppola N., et al. Naturally occurring mutations associated with resistance to HCV NS5B polymerase and NS3 protease inhibitors in treatment-naïve patients with chronic hepatitis C. Virol. J. 2015; 12: 186. https://doi.org/10.1186/s12985-015-0414-1
- Iio E., Shimada N., Abe H., Atsukawa M., Yoshizawa K., Takaguchi K., et al. Efficacy of daclatasvir/asunaprevir according to resistance-associated variants in chronic hepatitis C with genotype 1. J. Gastroenterol. 2017; 52(1): 94–103. https://doi.org/10.1007/s00535-016-1225-x
- Spitz N., Barros J.J., do Ó K.M., Brandão-Mello C.E., Araujo N.M. The first complete genome sequences of hepatitis C virus subtype 2b from Latin America: molecular characterization and phylogeographic analysis. Viruses. 2019; 11(11): 1000. https://doi.org/10.3390/v11111000
- Bull R.A., Eltahla A.A., Rodrigo C., Koekkoek S.M., Walker M., Pirozyan M.R., et al. A method for near full-length amplification and sequencing for six hepatitis C virus genotypes. BMC Genomics. 2016; 17: 247. https://doi.org/10.1186/s12864-016-2575-8
- Aisyah D.N., Story A., Kremyda-Vlachou M., Kozlakidis Z., Shalcross L., Hayward A. Assessing hepatitis C virus distribution among vulnerable populations in London using whole genome sequencing: results from the TB-REACH study. WellcomeOpenRes. 2021; 6: 229. https://doi.org/10.12688/wellcomeopenres.16907.1
- Kotova V.O., Balakhontseva L.A., Bazykina E.A., Trotsenko O.E., Beldy V.N., Kirdyashova S.E. Genetic diversity of hepatitis c virus in Nanaian region, Khabarovsk territory. Infektsiya i immunitet. 2021; 11(1): 148–56. https://doi.org/10.15789/2220-7619-GDO-1265 https://elibrary.ru/enfery (in Russian)
- Reingardt D.E., Ostankova Yu.V., Lyalina L.V., Anufrieva E.V., Semenov A.V., Totolian A.A. Distribution of hepatitis С virus drug resistance mutations among patients with recurrence of the disease during therapy with direct antiviral drugs. VICh-infektsiya i immunosupressii. 2023; 15(4): 86–93. https://doi.org/10.22328/2077-9828-2023-15-4-86-93 (in Russian)
- Isakov V., Hedskog C., Wertheim J.O., Hostager R.E., Parhy B., Schneider A.B., et al. Prevalence of resistance-associated substitutions and phylogenetic analysis of hepatitis C virus infection in Russia. Int. J. Infect. Dis. 2021; 113: 36–42. https://doi.org/10.1016/j.ijid.2021.09.041
- Chanyshev M.D., Vlasenko N.V., Roev G.V., Kotov I.A., Glushchenko A.G., Makashova V.V., et al. NGS amplification panel for HBV (Hepadnaviridae: Orthohepadnavirus) sequencing. Voprosy virusologii. 2024; 69(1): 65–75. https://doi.org/10.36233/0507-4088-212 (in Russian)
- Cheng X.D., Xu H.F., Wei X.M., Zhou H.Z. Variation analysis of E1 and E2 in HCV subtypes. Arch. Virol. 2015; 160(10): 2479–82. https://doi.org/10.1007/s00705-015-2533-9
- Kandel S., Hartzell S.L., Ingold A.K., Turner G.A., Kennedy J.L., Ussery D.W. Genomic surveillance of SARS-CoV-2 using long-range PCR primers. Front. Microbiol. 2024; 15: 1272972. https://doi.org/10.3389/fmicb.2024.1272972
- Koskela von Sydow A., Lindqvist C.M., Asghar N., Johansson M., Sundqvist M., Mölling P., et al. Comparison of SARS-CoV-2 whole genome sequencing using tiled amplicon enrichment and bait hybridization. Sci. Rep. 2023; 13(1): 6461. https://doi.org/10.1038/s41598-023-33168-1
- Yoshimi S., Imamura M., Murakami E., Hiraga N., Tsuge M., Kawakami Y., et al. Long term persistence of NS5A inhibitor-resistant hepatitis C virus in patients who failed daclatasvir and asunaprevir therapy. J. Med. Virol. 2015; 87(11): 1913–20. https://doi.org/10.1002/jmv.24255
- Wyles D.L., Luetkemeyer A.F. Understanding hepatitis C virus drug resistance: clinical implications for current and future regimens. Top Antivir. Med. 2017; 25(3): 103–9.
Supplementary files

