



Амплификационная панель NGS HCV-seq для таргетного секвенирования РНК вируса гепатита С (Flaviviridae: Hepacivirus)
- Авторы: Чанышев М.Д.1, Чернышова А.С.1,2, Глущенко А.Г.1,2, Гришаева А.А.1, Макашова В.В.1, Понежева Ж.Б.1, Хафизов К.Ф.1, Акимкин В.Г.1
-
Учреждения:
- ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
- ФГАОУ ВО «Московский физико-технический институт (национальный исследовательский университет)»
- Выпуск: Том 70, № 4 (2025)
- Страницы: 363-373
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://virusjour.crie.ru/jour/article/view/16786
- DOI: https://doi.org/10.36233/0507-4088-331
- EDN: https://elibrary.ru/fbgobh
- ID: 16786

Цитировать

Аннотация
Введение. Гепатит С является актуальной проблемой здравоохранения во всем мире. Высокая вариабельность вируса гепатита С (ВГС) осложняет его полногеномное секвенирование, в большинстве исследований секвенированы только отдельные регионы генома. Существует необходимость в простом и надежном методе секвенирования полного генома ВГС.
Цель работы. Создание и апробация панели NGS для полногеномного секвенирования ВГС.
Материалы и методы. В работе представлена амплификационная панель NGS для секвенирования генома ВГС генотипов 1, 2 и 3. В зависимости от генотипа использован набор, состоящий из 79, 67 или 89 праймеров, при помощи которых осуществляется амплификация перекрывающихся участков генома ВГС.
Результаты. Панель была апробирована на 153 образцах РНК ВГC, выделенных из крови (93/6/54 образца генотипов 1/2/3 соответственно). Анализ энтропии Шеннона показал, что в пределах гена E2 генетическая неоднородность значительно выше, чем в остальной части генома. Была определена частота встречаемости мутаций, ассоциированных с лекарственной резистентностью. В частности, для генотипа 1 была отмечена следующая частота встречаемости мутаций в NS3: Y56F – 37,6%, V170I – 23,7%; в NS5a: R30Q – 8,6%, P58L/S/T – 6,5%, A92T – 4,3%; в NS5b: L159F – 45,2%, S556G/N – 33,3%.
Заключение. В работе изложен способ полногеномного секвенирования генома ВГС генотипов 1, 2 и 3. Панель для секвенирования ВГС обладает большим потенциалом для использования в научных исследованиях и эпидемиологическом мониторинге.
Ключевые слова
Полный текст
Введение
Вирусный гепатит С представляет собой серьезную проблему для здравоохранения как в России, так и во всем мире. По оценкам Всемирной организации здравоохранения, в 2022 г. в мире было зарегистрировано около 50 млн человек, страдающих хроническим гепатитом С (ХГС), и 1,0 млн новых случаев инфицирования. В 2020 г. в России было зарегистрировано 24,5 тыс. пациентов с диагнозом ХГС, а уровень заболеваемости составил 16,7 на 100 тыс. человек [1]. По состоянию на 2024 г. предположительно 2,9% населения Российской Федерации инфицированы вирусом гепатита С (ВГС). В среднем у 70% инфицированных развивается хроническая форма заболевания, способная привести к циррозу печени (ЦП) и гепатоцеллюлярной карциноме (ГЦК). Согласно результатам анализа заболеваемости, распространенности и смертности ВГС, в 2015 г. было зарегистрировано 16 427 летальных исходов в результате осложнений ХГС в России [2].
Возбудителем заболевания является РНК-содержащий вирус из семейства Flaviviridae (Flaviviridae: Hepacivirus: Hepacivirus hominis). Геном ВГС представлен одноцепочечной РНК и демонстрирует высокую вариабельность. Известно 7 основных генотипов ВГС, различия нуклеотидных последовательностей между генотипами составляют 30–35%. Частота встречаемости генотипов отличается для разных регионов мира: генотип 1 преобладает в Европе, Северной и Южной Америке, генотип 3 – в Индии и ряде стран Юго-Восточной Азии. На территории РФ практически все типированные образцы относятся к генотипу 1 (примерно 70% случаев), 2 (~ 5%) и 3 (~ 20%) [3, 4].
Многочисленные исследования показывают, что различные генетические варианты ВГС и мутации в разных участках вирусного генома могут быть связаны с клиническими особенностями течения заболевания и его исходами, а также резистентностью к терапии. Например, отмечалось, что генотип 1b характеризовался высоким риском ЦП и ГЦК [5]. Аминокислотные замены, обуславливающие лекарственную устойчивость ВГС, расположены преимущественно в генах, кодирующих вирусные ферменты, таких как NS3, NS5a и NS5b [6]. Устойчивые к ВГС варианты встречаются у большинства пациентов, не достигших устойчивого вирусологического ответа, из-за отбора и роста устойчивых к ВГС вариантов в организме конкретного носителя. Эти мутации, связанные с устойчивостью, зависят от класса используемых противовирусных препаратов прямого действия (ПППД), а также варьируются в зависимости от генотипа и подтипа ВГС [7]. Представляют интерес и другие регионы вирусного генома. Делеция в NS2 может быть ассоциирована с ЦП [8]. В исследовании S. Fishman и соавт. мутации в core 36G/C, 209A, 271U/C, 309A/C, 435A/C, 481A и 546A/C были связаны с высоким риском ГЦК [9].
В большинстве исследований, включающих секвенирование РНК ВГС, определяли отдельные регионы вирусного генома [9–11]. На настоящий момент опубликовано небольшое число работ, в которых был определен полный геном ВГС, что может быть связано с высокой генетической вариабельностью этого вируса. Так, одна из работ за 2019 г. включала всего 2 образца и представляла собой первые полные прочтения ВГС на территории Латинской Америки. В ходе работы был использован метод вложенной полимеразной цепной реакции (ПЦР) (nested PCR) c последующим секвенированием амплифицированных участков по Сэнгеру [12]. Похожий подход для амплификации был использован в работе для сравнения чувствительности секвенирования полногеномных геномов ВГС на платформах PacBio и Illumina для обнаружения низкочастотных мутаций [13]. Аналогичная пробоподготовка и секвенирование на Illumina MiSeq были произведены для 88 образцов с целью получения данных о распределении генотипов в лондонской выборке [14].
России секвенирование ВГС проводится преимущественно для определения вирусного генотипа, анализируются лишь отдельные гены ферментов методом секвенирования по Сэнгеру. Например, В.О. Котова и соавт. определяли нуклеотидную последовательность гена NS5b в 60 образцах ВГС [15]. В работе Д.Э. Рейнгардт и соавт. были секвенированы гены NS3, NS5a, NS5b в 31 образце [16]. В.А. Исаков и соавт. анализировали встречаемость замен в генах NS3, NS5A, NS5B, ассоциированных с лекарственной устойчивостью, в образцах российской популяции, при этом секвенирование на базе Illumina MiSeq выполнялось в США [17]. По нашим оценкам, на сегодняшний день в мире существует крайне ограниченное число работ, в которых авторы секвенировали полный геном ВГС с помощью NGS. На территории Российской Федерации такие публикации нами не были обнаружены.
Цель работы – разработать амплификационную панель NGS для полногеномного секвенирования ВГС генотипов 1, 2 и 3 и провести ее тестирование.
Материалы и методы
Образцы
Для работы были отобраны 153 образца плазмы крови, полученных от участников клинического исследования (протокол № 133 от 2 марта 2023 г. ЛЭК ЦНИИ Эпидемиологии Роспотребнадзора) с ВГС. Все участники исследования предоставили добровольное информированное согласие на использование образцов. РНК ВГС выделяли из плазмы при помощи набора «РИБО-преп» («АмплиСенс», Россия). Наличие РНК ВГС и генотип определяли с использованием набора реагентов «HCV-Monitor-FL» и «АмплиСенс HCV-1/2/3-FL» («АмплиСенс», Россия). Из них 93 образца были определены как генотип 1, 6 – как генотип 2, 54 – как генотип 3.
Подбор праймеров
Выполненный биоинформатический анализ показал невозможность подбора универсальных праймерных последовательностей для полногеномной амплификации ВГС одновременно для всех трех основных генотипов вируса, циркулирующих в России. В то же время возможно подобрать три набора праймеров, которые были бы применимы для каждого из трех генотипов по отдельности. Для каждого генотипа праймеры подбирали в полуавтоматическом режиме, как было описано ранее для разработки амплификационной панели для секвенирования генома ВГС [18]. В результате были подобраны 79/67/89 праймеров для генотипов 1/2/3 соответственно. Три контрольных образца, соответствующие генотипам 1, 2 и 3 ВГС (HCV1_01, HCV2_01, HCV3_01), были последовательно секвенированы несколько раз, в результате были подобраны оптимальные условия ПЦР и концентрация каждого праймера в реакционной смеси. Праймерные последовательности приведены в табл. S1, представленной в дополнительных материалах.
Секвенирование ВГС
Для обратной транскрипции использовали набор «РЕВЕРТА-L» («АмплиСенс», Россия). Для амплификации генома ВГC использовали три набора праймеров, каждый соответствовал генотипу 1, 2 или 3. Генотипы образцов были определены заранее. Для каждого образца осуществляли 5 отдельных мультиплексных ПЦР, каждая реакция содержала 10 мкл комплементарной ДНК (кДНК), 10 мкл смеси для ПЦР PCR-mix-2-blue («АмплиСенс», Россия), 1,4 мкл dNTP 4,4 мМ («АмплиСенс», Россия), праймеры одного из 5 пулов и воду mQ в конечном объеме 25 мкл. Таблица S1 в дополнительных материалах содержит информацию о нуклеотидной последовательности, концентрации в реакционной смеси и номере пула для каждого праймера. Стадии ПЦР: 1) 95 °C 3 мин; 2) 20 циклов: 95 °C 30 с, 55 °C 30 с, 72 °C 40 с; 3) 72 °C 3 мин. Далее продукты 5 параллельных реакций ПЦР смешивали по 5 мкл и производили их очистку при помощи AMPure XP Beads (Beckman Coulter, США), соотношение AMPure XP Beads/продукт ПЦР как 0,8 : 1. Затем осуществляли ПЦР-индексацию, каждая реакция содержала 5 мкл очищенных продуктов ПЦР, 10 мкл смеси для ПЦР PCR-mix-2-blue («АмплиСенс», Россия), 1,4 мкл dNTP 4,4 мМ («АмплиСенс», Россия), стандартные адаптеры Nextera index (концентрация в реакционной смеси 200 нМ) и воду mQ в конечном объеме 25 мкл. Стадии ПЦР: 1) 95 °C 3 мин; 2) 25 циклов: 95 °C 20 с, 55 °C 30 с, 72 °C 40 с; 3) 72 °C 3 мин. Индексированные продукты ПЦР смешивали и далее производили их очистку при помощи AMPure XP Beads 0,8/1. Для измерения концентрации полученной библиотеки использовали Qubit dsDNA HS Assay Kit (Invitrogen, США). Секвенирование осуществляли с использованием набора MiSeq v. 3 (600 циклов) на Illumina MiSeq.
Биоинформатика
Для оценки качества полученных прочтений использовали программу FastQC. Адаптерные последовательности удаляли с помощью программы Trimmomatic. Для удаления праймерных последовательностей применяли программу Cutadapt. Обработанные таким образом прочтения картировали на соответствующие референсы (M58335.1/NC_009823.1/D17763.1 для генотипов 1/2/3) при помощи программы BWA-MEM. Консенсусные последовательности были получены из bam-файлов с использованием iVar. Для построения филогенетического древа были использованы 42 референсных генома ВГС из базы данных euHCVdb, представляющих генотипы с 1-го по 7-й. Множественное выравнивание последовательностей производили с использованием MAFFT v. 7.505. Филогенетическое дерево было построено в MEGA v. 12.0.11 с использованием алгоритма присоединения соседей и укоренено при помощи метода средней точки. Визуализация выполнена с использованием программы iTOL.
Валидация панели
Для валидации HCV-seq секвенировали три контрольных образца разных генотипов (HCV1_01, HCV2_01, HCV3_01), используя альтернативный подход. Обратную транскрипцию проводили при помощи набора SuperScript IV Reverse Transcriptase (ThermoFisher, США) согласно рекомендациям производителя. Для амплификации РНК ВГС для каждого образца использовали отдельный набор праймеров (приведены в табл. S2 в дополнительных материалах). ПЦР-смесь содержала 10 мкл матричной кДНК, 12,5 мкл Q5 High-Fidelity DNA Polymerase MasterMix 2x (NEB, США), праймеры в конечной концентрации 200 нМ и воду mQ в конечном объеме 25 мкл. Стадии ПЦР: 1) 98 °C 1 мин; 2) 40 циклов: 98 °C 10 с, 60 °C 30 с, 72 °C 2 мин; 3) 72 °C 3 мин. Продукты ПЦР были проверены при помощи электрофореза в 1% агарозном геле. Очистку продуктов ПЦР производили с использованием AMPure XP Beads в соотношении 0,5 : 1. Концентраци. продуктов ПЦР определяли при помощи dsDNA HS Assay Kit (Invitrogen, США). Далее для каждого образца продукты ПЦР разводили до 1 нг/мкл и смешивали эквимолярно. Библиотеки для секвенирования готовили при помощи набора Nextera XT DNA (Illumina) согласно рекомендациям производителя. Секвенирование на платформе Illumina и сборку геномов осуществляли, как описано выше. Кроме того, для образца HCV2_01 (генотип 2) геномная последовательность была также подтверждена секвенированием по Сэнгеру. Результаты секвенирования образцов при помощи HCV-seq, Nextera и по Сэнгеру совпадали практически полностью. Файлы сырых прочтений NGS при помощи HCV-seq и Nextera для всех трех образцов, а также данные секвенирования по Сэнгеру образца HCV2_01 могут быть загружены по https://doi.org/10.5281/zenodo.15846624 ссылке.
Результаты
В рамках настоящей работы было секвенировано и проанализировано 153 генома ВГС. Полученные геномы представлены в виде HCV_SAMPLES.fasta по https://doi.org/10.5281/zenodo.15846624 ссылке. Среднее покрытие вирусного генома составило 96,2% от области, ограниченной праймерами (рис. 1). Глубина прочтения ампликонов для трех генотипов представлена на рис. 2. Неполное покрытие образцов может быть связано с высокой вариабельностью ВГС и наличием мутаций на местах посадки праймеров, а также с недостаточным количеством и качеством РНК ВГС.

Рис. 1. Покрытие 153 образцов.
Область генома считали прочитанной при глубине не менее 10 прочтений. Процент покрытия считали от области, ограниченной праймерами. Образцы отсортированы по проценту покрытия и генотипу.
Fig. 1. Coverage of 153 samples.
The genome region was considered as read when coverage was at least 10 reads. The coverage percentage was calculated for the region flanked by primers. Samples are sorted by coverage percentage and genotype.

Рис. 2. Log10 глубины прочтения для каждого ампликона, нормированной на общее количество прочтений на образец.
а – генотип 1, б – генотип 2, в – генотип 3. По оси X отмечены ампликоны, по оси Y – образцы. Непрочитанные (желтые) участки единично распределены по геномам и образцам и связаны с множественными мутациями на местах отжига праймера в конкретном образце.
Fig. 2. Log10 of the reads per amplicon normalized by total reads per sample.
a – genotype 1, b – genotype 2, c – genotype 3. The X-axis shows the amplicons, the Y-axis shows the samples. Unread (yellow) regions, singly distributed across genomes and samples, are associated with multiple mutations at the primer annealing sites in a given samples.
Филогенетический анализ (рис. 3) подтвердил точное разделение всех исследованных образцов по генотипам: образцы генотипа 1 (красный), генотипа 2 (синий) и генотипа 3 (зеленый) образовали устойчивые, хорошо различимые кластеры. Все секвенированные изоляты достоверно группировались с референсными последовательностями своих генотипов, что подтверждает корректность предварительного генотипирования. При этом наблюдалась четкая кластеризация по субтипам: генотип 1 с подтипом 1b, генотип 2 с подтипами 2a и 2c, а генотип 3 с подтипом 3a.

Рис. 3. Филогенетическое дерево секвенированных образцов ВГC.
Цвета генотипов соответствуют [3]. Референсные последовательности отмечены черным цветом, а секвенированные в данной работе – синим.
Fig. 3. Phylogenetic tree of sequenced HCV samples.
The genotype colors correspond to [3]. Reference sequences are labeled in black, while sequences obtained in this study are highlighted in blue.
Для оценки нуклеотидной вариабельности был проведен анализ энтропии Шеннона, отражающей степень неопределенности в распределении нуклеотидов по позициям. На рис. 4 показаны средние значения энтропии Шеннона, рассчитанные для каждого генотипа по неперекрывающимся блокам длиной 40 нуклеотидов. Статистический анализ показал, что среднее значение энтропии Шеннона в пределах гена E2 значительно больше, чем в остальной части генома во всех трех проанализированных генотипах ВГС (p < 0,0001 по критерию Манна–Уитни). Это подтверждает наблюдаемый пик вариабельности в данной области особенно в 5’-регионе гена E2. Повышенная энтропия в регионах E1 и особенно E2 отражает адаптацию вируса к иммунному давлению. Известно, что область 5’-конца гена E2 кодирует N-концевой гипервариабельный регион HVR1, который связан с ускользанием от нейтрализующих антител и взаимодействием с рецепторами клетки-хозяина [19].

Рис. 4. Среднее значение энтропии Шеннона для неперекрывающихся участков генома длиной 40 нуклеотидов.
a – профиль энтропии Шеннона для генотипа 1, б – профиль энтропии Шеннона для генотипа 2, в – профиль энтропии Шеннона для генотипа 3, D – схематическое представление генома ВГС с обозначением границ генов. Пунктирные линии обозначают границы генов E1 и E2.
Fig. 4. Average Shannon entropy for non-overlapping 40-nucleotide regions of the genome.
a – Shannon entropy profile for genotype 1, b – Shannon entropy profile for genotype 2, c – Shannon entropy profile for genotype 3. D – Schematic representation of the HCV genome with genome region boundaries indicated. Dashed lines indicate the boundaries of the E1 and E2 regions.
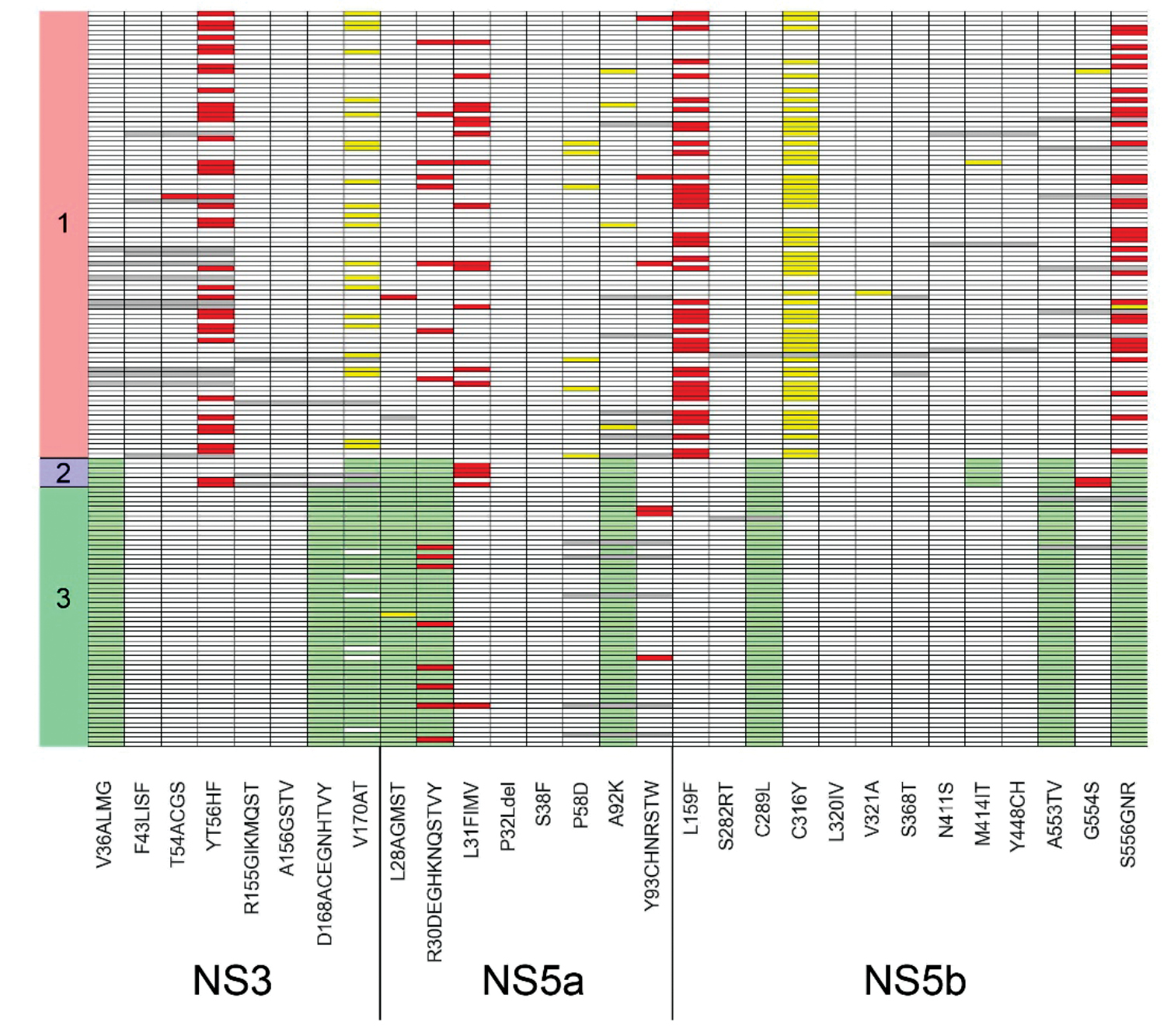
Далее была произведена оценка встречаемости мутаций ВГС, связанных с лекарственной резистентностью. Соответствие мутаций и препаратов осуществлялось согласно работе1. Обнаруженные мутации представлены в виде диаграммы на рис. 5. Для генотипа 1 была отмечена следующая частота встречаемости мутаций в NS3: Y56F – 37,6%, V170I – 23,7%; в NS5a: R30Q – 8,6%, P58L/S/T – 6,5%, A92T – 4,3%; в NS5b: L159F – 45,2%, S556G/N – 33,3%. Для образцов генотипа 2 были отмечены характерные для данного генотипа мутации – V36L и V170I в NS3, L28С/F, R30K и A92C/S в NS5a и C289M, M414Q, A553V и S556G в NS5b. Кроме того, встречались замены Y56F в NS3, L31M в NS5a и G554S в NS5b. Для образцов генотипа 3 также были выявлены характерные для данного генотипа мутации: V36L, D168Q и V170I в NS3, L28M, R30A и A92E в NS5a, C289F, A553V и S556G в NS5b. Для генотипа 3 была отмечена следующая частота встречаемости мутаций в NS5a: R30K/S/T – 14,8%, L31I – 1,9% и Y93H – 5,6%. Во всех образцах не было обнаружено мутаций резистентности к препаратам группы ингибиторов NS3/4A глекапревиру и паритапревиру. В то же время образцы ВГС генотипа 1 содержали возможные мутации резистентности к препаратам группы ингибиторов NS5A (деклатасвир, элбасвир, ледипасвир, велпатасвир), которые обнаружены в 95,7% образцов, а для группы ингибиторов NS5B (досабувир и софасбурив) частота мутаций составила 45,1%. Образцы ВГС генотипа 2 содержали возможные мутации резистентности к препаратам группы ингибиторов NS5A в 66,7% образцов, а для препаратов группы ингибиторов NS5B не были обнаружены мутации резистентности. Образцы ВГС генотипа 3 содержали возможные мутации резистентности к препаратам группы ингибиторов NS5A в 15,1% образцов, а для препаратов группы ингибиторов NS5B мутации резистентности не определялись.

Рис. 5. Встречаемость мутаций в генах NS3, NS5a, NS5b, ассоциированных с резистентностью к терапии.
Серые поля представляют собой неопределенные области, белые – дикий тип, зеленые – мутации, характерные для данного генотипа, желтые – замены, не описанные в литературе, красные – мутации, описанные в литературе как мутации устойчивости к противовирусной терапии. По оси X отмечены мутации и гены, по оси Y – образцы и генотипы.
Fig. 5. The detection rates of mutations in the NS3, NS5a, and NS5b coding regions associated with resistance to therapy.
Gray fields represent undefined areas, white – wild type, green – mutations characteristic of the genotype, yellow – substitutions not described in the literature, and red – mutations described in the literature as mutations of resistance to antiviral therapy. The X-axis shows mutations and genome regions, the Y-axis shows samples and genotypes.
Обсуждение
В настоящей работе представлен метод полногеномного секвенирования ВГС, основанный на ПЦР-обогащении сравнительно коротких ампликонов. Существует небольшое количество работ, включающих полногеномное секвенирование ВГС. В работе N. Spitz и соавт. была использована вложенная ПЦР, длина ампликонов составляла примерно 4,5 т.п.н. Далее полученные ампликоны были секвенированы по Сэнгеру с использованием внутренних праймеров [12]. В исследованиях R. Bull и соавт. и D. Aisyah и соавт. праймеры были подобраны так, чтобы амплифицировать полный вирусный геном, ампликоны далее тагментировали и секвенировали на платформе Illumina [13, 14]. Следует отметить, что данный подход подразумевает получение протяженных ампликонов 4–9 т.п.н., что требует наличия достаточного количества полноразмерных копий вирусного генома в образце, в то время как многие образцы содержат фрагментированную РНК. Использование коротких ампликонов позволяет секвенировать большую часть клинических образцов. Интересно отметить, что для рутинного секвенирования миллионов геномов SARS-CoV-2 в мире использовали способы с амплификацией сравнительно коротких участков вирусного генома ARTIC и Midnight [20, 21]. Представленный в настоящей работе способ также имеет недостатки. Независимая амплификация коротких участков вирусного генома не позволяет детектировать протяженные делеции фракции вирусных геномов, которые были отмечены в работе K. Yamauchi и соавт. [8].
Полученные в настоящем исследовании данные о генотипическом разнообразии ВГС в выборке российской популяции в целом согласуются с немногочисленными опубликованными результатами других научных групп. Например, в исследовании Н.Н. Пименова и соавт., проведенном на 10 107 образцах, частота встречаемости генотипов ВГС 1/2/3 на территории РФ составила 53,6, 7,6 и 35,4% случаев соответственно [4]. Частота встречаемости генотипов 1/2/3 в нашей относительно небольшой выборке из 153 образцов составила 60,8/3,9/35,3% случаев. Д.Э. Рейнгардт и соавт. определяли нуклеотидную последовательность генов NS3, NS5a, NS5b в 31 образце [16]. В целом набор и частота обнаруженных мутаций в их исследовании соотносятся с полученными нами результатами. Для ряда мутаций частота встречаемости в работе Д.Э. Рейнгардт и соавт. была значительно больше. Возможно, это связано с тем, что материалом исследования служил 31 образец плазмы крови от пациентов с ХГС с рецидивом заболевания на фоне терапии ПППД, в то время как значительную часть нашей выборки составляли пациенты с впервые выявленным ВГС. Например, частота встречаемости NS5a Y93H составила 3,9% в настоящем исследовании и 41,9% в работе Д.Э. Рейнгардт и соавт.
В настоящем исследовании наибольшая частота мутаций резистентности к терапии определялась к препаратам из группы ингибиторов белка NS5A ВГС. Данное наблюдение согласуется с результатами многочисленных опубликованных исследований. Так, известно, что ингибиторы белка NS5A ВГС наиболее подвержены формированию мутаций резистентности. Исследования показывают, что мутации в гене NS5A встречаются у значительной части пациентов (до 30% у наивных пациентов с генотипом 1b) [22]. Также следует отметить значительную перекрестную резистентность между доступными в настоящее время ингибиторами NS5A [23]. Мутации резистентности к ингибиторам NS5A и NS3 часто накапливаются у пациентов без устойчивого вирусологического ответа после лечения с применением препаратов этих групп. В то же время ингибиторы NS3/4A и NS5B также используются в терапии, но частота мутаций резистентности к ним ниже. Например, ингибитор NS5B софосбувир обладает самым высоким барьером к резистентности и мутации к нему выявляются крайне редко. Ингибиторы NS3/4A могут вызывать мутации резистентности, но они менее устойчивы по сравнению с NS5A-ингибиторами [22]. Мутации резистентности к ингибиторам NS5B, по данным разных источников, обнаруживаются редко (1% случаев) даже после применения неэффективного режима ПППД, содержащего препараты из данной группы[2]. В исследованных нами образцах не было обнаружено мутаций резистентности к препаратам из группы ингибиторов NS3/4A, мутации резистентности к препаратам из группы ингибиторов NS5B встречались только среди пациентов с генотипом 1 ВГС. Таким образом, интеграция тестирования на лекарственную устойчивость в клиническую практику может повысить эффективность лечения гепатита С. Тестирование на устойчивость, в особенности к ингибиторам NS5A, может быть рекомендовано всем пациентам, не достигшим устойчивого вирусологического ответа после применения схем терапии, содержащих препараты данной группы.
Заключение
В настоящей работе представлена амплификационная панель, позволяющая осуществлять полногеномное секвенирование генотипов 1, 2 и 3 ВГС. Панель основана на амплификации относительно коротких участков вирусного генома, что позволяет секвенировать даже высокофрагментированные образцы, и обладает большим потенциалом для научных и практических целей. Широкое применение комбинированных безинтерфероновых схем позволило достигнуть существенных успехов в лечении ВГС. Несмотря на то что ПППД обеспечивают устойчивый вирусологический ответ более чем в 90% случаев, проблема лекарственной устойчивости становится все более актуальной. Высокая скорость размножения ВГС и низкая точность работы его РНК-полимеразы приводят к значительной генетической изменчивости вирусной популяции, что способствует быстрому появлению и отбору вариантов, устойчивых к терапии. Мониторинг лекарственной устойчивости ВГС имеет важное значение для успешного лечения пациентов с ХГС. Определение мутаций, вызывающих резистентность к ПППД, позволяет подобрать наиболее эффективную схему терапии и значительно повысить шансы пациента на полное излечение.
2 AASLD. HCV Resistance Primer. HСV Guidance; 2022. Available at: https://hcvguidelines.org/evaluate/resistance
Об авторах
Михаил Дамирович Чанышев
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Автор, ответственный за переписку.
Email: chanishq@gmail.com
ORCID iD: 0000-0002-6943-2915
канд. биол. наук, старший научный сотрудник Лаборатории геномных исследований
Россия, 111123, г. МоскваАнастасия Сергеевна Чернышова
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора; ФГАОУ ВО «Московский физико-технический институт (национальный исследовательский университет)»
Email: chernyshova.a@cmd.su
ORCID iD: 0009-0000-2440-1309
лаборант Лаборатории геномных исследований ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. Москва; 141701, г. ДолгопрудныйАльбина Григорьевна Глущенко
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора; ФГАОУ ВО «Московский физико-технический институт (национальный исследовательский университет)»
Email: albinagluschenko@gmail.com
ORCID iD: 0009-0002-8851-8703
лаборант-исследователь Лаборатории геномных исследований ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. Москва; 141701, г. ДолгопрудныйАнтонина Алексеевна Гришаева
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: antoninagrishaeva@yandex.ru
ORCID iD: 0000-0002-1326-9274
канд. мед. наук, научный сотрудник Клинического отдела инфекционной патологии
Россия, 111123, г. МоскваВера Васильевна Макашова
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: veramakashova@yandex.ru
ORCID iD: 0000-0002-0982-3527
д-р мед. наук, ведущий научный сотрудник Клинического отдела инфекционной патологии
Россия, 111123, г. МоскваЖанна Бетовна Понежева
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: doktorim@mail.ru
ORCID iD: 0000-0002-6539-4878
д-р мед. наук, доцент, заведующая Клиническим отделом инфекционной патологии
Россия, 111123, г. МоскваКамиль Фаридович Хафизов
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: kkhafizov@gmail.com
ORCID iD: 0000-0001-5524-0296
канд. биол. наук, заведующий Лабораторией геномных исследований
Россия, 111123, г. МоскваВасилий Геннадьевич Акимкин
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
академик РАН, доктор медицинских наук, профессор, директор
Россия, 111123, г. МоскваСписок литературы
- Isakov V., Nikityuk D. Elimination of HCV in Russia: Barriers and perspective. Viruses. 2022; 14(4): 790. https://doi.org/10.3390/v14040790
- Пименов Н.Н., Комарова С.В., Карандашова И.В., Цапкова Н.Н., Волчкова Е.В., Чуланов В.П. Гепатит С и его исходы в России: анализ заболеваемости, распространенности и смертности до начала программы ликвидации инфекции. Инфекционные болезни. 2018; 16(3): 37–45. https://doi.org/10.20953/1729-9225-2018-3-37-45 https://elibrary.ru/ysjrqt
- Messina J.P., Humphreys I., Flaxman A., Brown A., Cooke G.S., Pybus O.G., et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015; 61(1): 77–87. https://doi.org/10.1002/hep.27259
- Pimenov N., Kostyushev D., Komarova S., Fomicheva A., Urtikov A., Belaia O., et al. Epidemiology and genotype distribution of hepatitis C virus in Russia. Pathogens. 2022; 11(12): 1482. https://doi.org/10.3390/pathogens11121482
- Raimondi S., Bruno S., Mondelli M.U., Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J. Hepatol. 2009; 50(6): 1142–54. https://doi.org/10.1016/j.jhep.2009.01.019
- Kalaghatgi P., Sikorski A.M., Knops E., Rupp D., Sierra S. Geno2pheno[HCV] – a Web-based interpretation system to support hepatitis C treatment decisions in the era of direct-acting antiviral agents. PLoS One. 2016; 11(5): e0155869. https://doi.org/10.1371/journal.pone.0155869
- Ahmed A., Felmlee D.J. Mechanisms of hepatitis C viral resistance to direct acting antivirals. Viruses. 2015; 7(12): 6716–29. https://doi.org/10.3390/v7122968
- Yamauchi K., Maekawa S., Osawa L., Komiyama Y., Nakakuki N., Takada H., et al. Single-molecule sequencing of the whole HCV genome revealed envelope deletions in decompensated cirrhosis associated with NS2 and NS5A mutations. J. Gastroenterol. 2024; 59(11): 1021–36. https://doi.org/10.1007/s00535-024-02146-3
- Fishman S.L., Factor S.H., Balestrieri C., Fan X., Dibisceglie A.M., Desai S.M., et al. Mutations in the hepatitis C virus core gene are associated with advanced liver disease and hepatocellular carcinoma. Clin. Cancer Res. 2009; 15(9): 3205–13. https://doi.org/10.1158/1078-0432.ccr-08-2418
- Costantino A., Spada E., Equestre M., Bruni R., Tritarelli E., Coppola N., et al. Naturally occurring mutations associated with resistance to HCV NS5B polymerase and NS3 protease inhibitors in treatment-naïve patients with chronic hepatitis C. Virol. J. 2015; 12: 186. https://doi.org/10.1186/s12985-015-0414-1
- Iio E., Shimada N., Abe H., Atsukawa M., Yoshizawa K., Takaguchi K., et al. Efficacy of daclatasvir/asunaprevir according to resistance-associated variants in chronic hepatitis C with genotype 1. J. Gastroenterol. 2017; 52(1): 94–103. https://doi.org/10.1007/s00535-016-1225-x
- Spitz N., Barros J.J., do Ó K.M., Brandão-Mello C.E., Araujo N.M. The first complete genome sequences of hepatitis C virus subtype 2b from Latin America: molecular characterization and phylogeographic analysis. Viruses. 2019; 11(11): 1000. https://doi.org/10.3390/v11111000
- Bull R.A., Eltahla A.A., Rodrigo C., Koekkoek S.M., Walker M., Pirozyan M.R., et al. A method for near full-length amplification and sequencing for six hepatitis C virus genotypes. BMC Genomics. 2016; 17: 247. https://doi.org/10.1186/s12864-016-2575-8
- Aisyah D.N., Story A., Kremyda-Vlachou M., Kozlakidis Z., Shalcross L., Hayward A. Assessing hepatitis C virus distribution among vulnerable populations in London using whole genome sequencing: results from the TB-REACH study. WellcomeOpenRes. 2021; 6: 229. https://doi.org/10.12688/wellcomeopenres.16907.1
- Котова В.О., Балахонцева Л.А., Базыкина Е.А., Троценко О.Е., Бельды В.Н., Кирдяшова С.Е. Генетическое разнообразие вируса гепатита С среди населения Нанайского района Хабаровского края. Инфекция и иммунитет. 2021; 11(1): 148–56. https://doi.org/10.15789/2220-7619-GDO-1265 https://elibrary.ru/enfery
- Рейнгардт Д.Э., Останкова Ю.В., Лялина Л.В., Ануфриева Е.В., Семенов А.В., Тотолян А.А. Распространенность мутаций лекарственной устойчивости вируса гепатита С среди пациентов с рецидивом заболевания на терапии препаратами прямого противовирусного действия. ВИЧ-инфекция и иммуносупрессии. 2023; 15(4): 86–93. https://doi.org/10.22328/2077-9828-2023-15-4-86-93
- Isakov V., Hedskog C., Wertheim J.O., Hostager R.E., Parhy B., Schneider A.B., et al. Prevalence of resistance-associated substitutions and phylogenetic analysis of hepatitis C virus infection in Russia. Int. J. Infect. Dis. 2021; 113: 36–42. https://doi.org/10.1016/j.ijid.2021.09.041
- Чанышев М.Д., Власенко Н.В., Роев Г.В., Котов И.А., Глущенко А.Г., Макашова В.В. и др. Амплификационная панель NGS для секвенирования ДНК вируса гепатита В (Hepadnaviridae: Orthohepa DNA virus). Вопросы вирусологии. 2024; 69(1): 65–75. https://doi.org/10.36233/0507-4088-212
- Cheng X.D., Xu H.F., Wei X.M., Zhou H.Z. Variation analysis of E1 and E2 in HCV subtypes. Arch. Virol. 2015; 160(10): 2479–82. https://doi.org/10.1007/s00705-015-2533-9
- Kandel S., Hartzell S.L., Ingold A.K., Turner G.A., Kennedy J.L., Ussery D.W. Genomic surveillance of SARS-CoV-2 using long-range PCR primers. Front. Microbiol. 2024; 15: 1272972. https://doi.org/10.3389/fmicb.2024.1272972
- Koskela von Sydow A., Lindqvist C.M., Asghar N., Johansson M., Sundqvist M., Mölling P., et al. Comparison of SARS-CoV-2 whole genome sequencing using tiled amplicon enrichment and bait hybridization. Sci. Rep. 2023; 13(1): 6461. https://doi.org/10.1038/s41598-023-33168-1
- Yoshimi S., Imamura M., Murakami E., Hiraga N., Tsuge M., Kawakami Y., et al. Long term persistence of NS5A inhibitor-resistant hepatitis C virus in patients who failed daclatasvir and asunaprevir therapy. J. Med. Virol. 2015; 87(11): 1913–20. https://doi.org/10.1002/jmv.24255
- Wyles D.L., Luetkemeyer A.F. Understanding hepatitis C virus drug resistance: clinical implications for current and future regimens. Top Antivir. Med. 2017; 25(3): 103–9.
Дополнительные файлы

