



The features of Tat protein of human immunodeficiency virus type 1 (Retroviridae: Lentivirus: Lentivirus humimdef1) non-A6 variants, characteristic for the Russian Federation
- Authors: Kuznetsova A.I.1, Antonova A.A.1, Lebedev A.V.1, Ozhmegova E.N.1, Shlykova A.V.2, Lapovok I.A.2, Galzitskaya O.V.1,3,4
-
Affiliations:
- D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya
- Central Research Institute of Epidemiology
- Institute of Protein Research RAS
- Institute of Theoretical and Experimental Biophysics RAS
- Issue: Vol 69, No 6 (2024)
- Pages: 524-534
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16690
- DOI: https://doi.org/10.36233/0507-4088-272
- EDN: https://elibrary.ru/xdvhfq
- ID: 16690

Cite item

Abstract
Introduction. Tat protein is a trans-activator of HIV-1 genome transcription, with additional functions including the ability to induce the chronic inflammatory process. Natural amino acid polymorphisms in Tat may affect its functional properties and the course of HIV infection.
The aim of this work is to analyze the marks of Tat consensus sequences in non-A6 HIV-1 variants characteristic of the Russian Federation, as well as study natural polymorphisms in Tat CRF63_02A6 and subtype B variants circulating in Russia.
Materials and methods. The whole-genome nucleotide sequences of HIV-1 CRF63_02A6, CRF03_A6B, as well as subtype B and CRF02_AG circulating in Russia were used. The reference group was formed based on the sequences of subtype B variants circulating in different countries. Preferentially, the sequences were downloaded from the international database Los Alamos.
Results. CRF63_02A6 consensus sequence contained the highest number of amino acid substitutions, 31, and had no helix at positions 30‒33 in the secondary structure; however, this did not change its predicted tertiary structure. CRF03_A6B consensus sequence contained a stop codon at position 87. The polymorphisms in subtype B variants circulating in our country and in CRF63_02A6 variants were identified.
Conclusion. Consensus sequences of Tat protein in non-A6 variants typical for the Russian Federation were obtained and their features were determined. R78G, located in the functionally significant motif, and C31S, the functionally significant substitution, were significantly more frequent in subtype B variants circulating in Russia and in CRF63_02A6 variants than in the reference group, respectively. A limitation of this study is the small sample of sequences.
Full Text
Introduction
Human immunodeficiency virus (HIV) belongs to the Lentivirus genus of the Orthoretrovirinae subfamily of the Retroviridae family. Based on genetic features and differences in viral antigens, HIV is classified into types 1 and 2 [1]. The spread of HIV-2 is limited to western Africa, although cases of importation of this type of virus into other parts of the world have been reported. HIV-1 originated around the 1920s in what is now the Democratic Republic of Congo and has spread across the world over time [2]. HIV-1 is categorized into groups based on genetic characteristics: M, N, O and P. Group M viruses account for the majority of HIV infections worldwide. Group M HIV-1 variants are divided into subtypes: A (sub-subtypes A1‒A8), B, C, D, F (sub-subtypes F1‒F2), G, H, J, K, L. Numerous recombinant forms are emerged between subtypes [3]. HIV-1 variants are spread worldwide very unevenly [2]. At the current stage of HIV-1 epidemic in Russia, sub-subtype A6 remains the dominant genetic variant (82.9%), subtype B (7.14%) ranks second in frequency of occurrence, the recombinant form CRF63_02A6 accounts for about 3.59%, the frequency of occurrence of each of the recombinant forms CRF02_AG and CRF03_AB is about 1% [4]. In the Russian Federation as a whole, the frequency of recombinant forms of HIV-1 and their involvement in the epidemic process has been increasing over time [4].
The Tat protein of HIV-1 is a trans-activator of viral genome transcription which alters the activity of the viral promoter and cellular RNA polymerase. Viral replication begins with transcription that results in the production of short viral RNAs encoding the Tat protein and several other viral proteins. The resulting transcripts are transported to the cytoplasm, where synthesis of the corresponding proteins occurs on ribosomes [5]. The newly formed Tat, possessing a nuclear localization signal, returns back to the nucleus, where it causes the release of positive transcription elongation factor b (P-TEFb) from the inactive complex formed by this protein with HEXIM1, LARP and 7SK RNA. Then P-TEFb in a complex with Tat binds to a special TAR element (trans-activation response element) on the synthesized viral RNA, which leads to an increase in the processivity of RNA polymerase and, as a consequence, to the formation of full-length viral RNA molecules [5, 6]. Tat also has additional intracellular and extracellular functions. Infected cells release Tat into the intercellular space, from where it enters the bloodstream. Afterwards, Tat protein can be taken up by both HIV-infected and uninfected cells. Latently HIV-infected cells can be reactivated by Tat protein. Uninfected cells that have engulfed the Tat protein enter become activated, which eventually leads them to apoptosis. Furthermore, the cells that have engulfed Tat themselves begin to produce inflammatory cytokines. As a result, a chronic inflammatory process is triggered, contributing to the development of comorbid, neurodegenerative and cardiovascular diseases in HIV-infected patients [5, 7, 8].
Tat is a small basic protein that is encoded by two exons and contains 86 to 106 amino acid residues (a.a.r.), predominantly 101 a.a.r. The first 5 domains are encoded by the first exon and the 6th domain is encoded by the second exon. The first 3 domains (1st: 1‒21 a.a.r.; 2nd: 22‒37 a.a.r.; 3rd: 38‒48 a.a.r.) form the minimal region required for trans-activation [6, 8]. The fourth domain (49‒57 a.a.r.) is responsible for binding to the TAR element, as well as protein uptake by cells and, together with the 5th domain (58‒72 a.a.r.), determines the nuclear localization of Tat [5, 6]. The sixth domain (73‒101 a.a.r.), encoded by the second exon, presumably contributes to viral infectivity and binding to integrins at the cell membrane [6]. The influence of amino acid substitutions in the Tat protein on its functions [9, 10] and on the pathogenesis of HIV infection [6, 11, 12] is being actively studied. The relevance of studying the variability of Tat protein is also determined by the fact that it is a promising target for the development of antiretroviral drugs and therapeutic vaccines [13, 14].
Previous studies of the Tat protein characteristics of the most prevalent sub-subtype A6 in Russia have identified substitutions that allow distinguishing this HIV variant from other genetic variants. Thus, studies have shown the presence of mutations in the 4th functionally significant domain, the frequency of which differed significantly between sub-subtype A6 and the most studied subtype B, and also identified the QRD motif in the 6th domain of the Tat protein in sub-subtype A6 instead of the functionally significant RGD motif [5, 7].
The aim of this study is to investigate the features of the Tat protein in non-A6 variants of HIV-1 characteristic of the Russian Federation: analysis of the features of consensus sequences of the Tat protein, including the study of secondary and tertiary structures, comparison of the profile of natural Tat polymorphisms in CRF63_02A6 variants and subtype B virus variants circulating in Russia with subtype B virus variants circulating worldwide. The data obtained can be used in the development of drugs and vaccines, as well as contribute to the study of the influence of polymorphisms on the functional properties of viruses.
Materials and methods
All whole-genome sequences of CRF63_02A6, CRF03_A6B variants, as well as subtype B and CRF02_AG variants circulating in the Russian Federation were selected from the Los Alamos international database (www.hiv.lanl.gov/content/index, dated April 19, 2024). One sequence from one patient was included in the study. The nucleotide sequences of the tat gene were retrieved from the selected sequences. As a result, 26 sequences of CRF63_02A6, 4 sequences of CRF03_A6B, 35 sequences of subtype B variants circulating in the territory of Russia, as well as one sequence of variant CRF02_AG obtained from an HIV-infected patient in Russia were downloaded.
Additionally, two sequences of the tat gene of the CRF02_AG variant, retrieved from whole-genome sequences of the virus obtained earlier by the laboratory from 2 patients within the framework of the CHAIN project of the 7th Framework Program of the European Community «Single Network for the Study of Drug Resistance to Antiretroviral Drugs», were included in the study. The Ethical Committee of the Federal State Unitary Scientific Center «Vector» obtained permission for blood collection from the patients (Protocol No. 1 of March 30, 2010). Patients signed an informed consent for participation in the study. Samples were analyzed by mass parallel sequencing using the AmpliSens HIV-Resist-NGS kit according to the manufacturer’s instructions (FBIS Central Research Institute for Disease Control of Rospotrebnadzor, Russia). Whole genome sequencing of samples was performed using MiSeq technology and appropriate MiSeq reagent kits V2 (Illumina, USA) by analyzing 4 overlapping specific fragments (total length of the analyzed fragment 704-9563 by HXB2).
The determination of the virus subtype was based on whole-genome sequence analysis in the Comet (https://comet.lih.lu) and RIP (RIP 3.0 submission form (lanl.gov)) programs. Sequences were grouped according to virus subtype.
Fifty whole-genome sequences of subtype B circulating in the USA, EU countries, Canada, Japan, China, South Korea and Australia were selected from the Los Alamos international database (www.hiv.lanl.gov/content/index) to form a reference group of sequences. One sequence from a single patient was also included in the study. From all the selected sequences, the nucleotide sequences of the tat gene were retrieved.
Nucleotide sequence quality control was then performed, in which the following sequences were excluded from analysis: a) those containing substitutions in the start codon; b) those containing nucleotide gaps not divisible by 3; and c) those containing 2 consecutive degenerate N positions. Sequences that failed quality control were removed from the study.
The nucleotide sequences were then translated into amino acid sequences using the Sequence Manipulation Suite: Translate program (www.bioinformatics.org) and aligned to each group in the MEGA v. 10.2.2 program (www.megasoftware.net). Next, an amino acid consensus sequence was generated for each sequence group using the Advanced Consensus Maker tool software on the Los Alamos database website (https://www.hiv.lanl.gov/content/sequence/CONSENSUS/AdvCon.html). Amino acid insertions were not taken into account when generating the reference sequence. A reference consensus sequence (reference) was generated based on a reference group of sequences.
Comparison of the consensus sequences CRF63_02A6, CRF03_A6B and CRF02_AG and subtype B variants circulating in Russia with the reference consensus sequence of subtype B and with each other was performed in the MEGA v. 10.2.2 program (www.megasoftware.net).
Further, the secondary structure of the consensus sequences of non-A6 variants of HIV-1 circulating in Russia was predicted based on consensus sequence analysis in the PSIPRED program (http://bioinf.cs.ucl.ac.uk/psipred/). Specific changes in the secondary structure of the corresponding virus variants relative to the reference consensus sequence were analyzed. The secondary structure was analyzed only for consensus sequences that were generated on the basis of more than 10 sequences.
The IsUnstruct program was used to predict the location of unstructured regions in consensus sequences [15].
The AlphaFold 2 program (AlphaFold Protein Structure Database) was used to predict the spatial structure of consensus sequences [16].
The natural polymorphisms of subtype B variants circulating in Russia and CRF63_02A6 variants were subsequently compared with subtype B variants circulating worldwide. For this purpose, initially, using the program MEGA v. 10.2.2 program, natural polymorphisms of all analyzed groups were detected relative to the reference consensus sequence – the consensus sequence of subtype B viruses circulating in the world. Polymorphisms were understood as mutations, i.e., single substitutions occurring in 1% of observations or more frequently [17]. Then, using the Nonparametric Statistics module of Statistica 8.0 (StatSoft Inc., USA), the group of subtype B variants circulating in Russia and the group of CRF63_02A6 variants were compared pairwise with the group of subtype B variants circulating worldwide: sites with statistically significant differences were identified (p < 0.05 using the χ2 criterion).
Results
Analyzed sequences
Two HIV-1 CRF02_AG whole-genome sequences obtained earlier during the CHAIN project were deposited in GeneBank under accession numbers PP816227 and PP816231.
After quality control of the downloaded nucleotide sequences, one CRF63_02A6 sequence, one CRF03_A6B sequence and two subtype B sequences from the reference group, i.e. the group of HIV-1 subtype B variant sequences circulating worldwide, were excluded from the analysis. Thus, the study included 25 sequences of CRF63_02A6, 3 sequences of CRF03_A6B, 35 sequences of subtype B variants and 3 sequences of CRF02_AG variant virus both circulating in the territory of the Russian Federation. The reference group was formed on the basis of 48 sequences. The obtained consensus sequences are presented in Fig. 1.

Fig. 1. Multiple alignment of the full-length Tat protein’s consensus sequences of subtype B and CRF02_AG variants circulating in Russia, and variants Crf03_A6B, CRF63_02A6 relative to the consensus sequence of subtype B variants circulating in the world (B reference).
The dots indicate amino acid residues (a.a.r.) positions in which the a.a.r. in the consensus corresponded to the reference. Non-polar amino acids: G (glycine), A (alanine), V (valine), L (leucine), I (isoleucine), P (proline) – are marked in blue; polar uncharged amino acids: S (serine), T (threonine), C (cysteine), M (methionine), N (asparagine), Q (glutamine) – green; aromatic amino acids: F (phenylalanine), Y (Tyrosine), W (tryptophan), H (Histidine) – yellow; polar acidic negatively charged amino acids: D (aspartic acid) and E (glutamic acid) – orange; polar basic positively charged amino acids: K (lysine), R (arginine) – in red [18, 19]. X – a.a.r. is undefined (gray).
Рис. 1. Множественное выравнивание консенсусных последовательностей полноразмерного белка Tat вариантов субтипа В и вариантов CRF02_AG, циркулирующих в России, и вариантов Crf03_A6B, CRF63_02A6 относительно консенсусной последовательности вариантов субтипа В, циркулирующих в мире (B референс).
Точками обозначены позиции АО, в которых АО в консенсусах соответствовали референсу. Аминокислоты классифицированы на основе полярности радикалов. Неполярные аминокислоты: G (глицин), A (аланин), V (валин), L (лейцин), I (изолейцин), P (пролин) отмечены синим цветом; полярные незаряженные аминокислоты: S (серин), T (треонин), C (цистеин), M (метионин), N (аспарагин), Q (глутамин) – зеленым; ароматические аминокислоты: F (фенилаланин), Y (тирозин), W (триптофан), H (гистидин) – желтым; отрицательно заряженные аминокислоты: D (аспарагиновая кислота) и E (глутаминовая кислота) – оранжевым; положительно заряженные аминокислоты: K (лизин), R (аргинин) – красным [18, 19]. X – АО не определен (серым).
Structural analysis
Primary structure
The consensus of HIV-1 subtype B variants circulating in Russia differed from the reference consensus sequence in 8 positions, and 8 of the 8 substitutions were associated with changes in chemical properties. A change in chemical properties was interpreted as a change in the polarity or charge of an amino acid at a specific position (Fig. 1).
The consensus of CRF02_AG virus variants circulating in Russia differed from the reference sequence in 30 positions, where the amino acid substitution was not associated with a change in chemical properties only in 8 out of the 30 positions.
The HIV-1 variant consensus of CRF03_A6B contained a premature stop codon at position 87, differed from the reference sequence in 6 of the 86 amino acid positions, where the amino acid substitution was not associated with a change in chemical properties in 3 of those 6 positions.
The consensus of CRF63_02A6 differed from the reference sequence in 31 positions, where the amino acid substitutions were not associated with a change in chemical properties in only 8 out of the 31 positions (Fig. 1).
Secondary structure
Secondary structure was investigated for consensus sequences, HIV-1 subtype B variants circulating in Russia, and CRF63_02A6 variants. They were compared with the reference sequence.
Since the consensus sequence of HIV-1 subtype B variants circulating in the Russian Federation was equally likely to contain serine (S) and proline (P) at position 70, two sequence variants were analyzed to predict the secondary structure of Tat protein: B(Russia)_v1/B(Russia)_v1 and B(Russia)_v2/B(Russia)_v2, respectively. The results of the structure analysis of the sequences under study are presented in Fig. 2.

Fig. 2. Predicted secondary structures of consensus sequences: A – B(референс)/B(reference); B – B(Россия)_v1/ B(Russia)_v1; C – B(Россия)_v2/ B(Russia)_v2; D – CRF63_02A6.
Рис. 2. Предсказанные вторичные структуры консенсусных последовательностей: A – B(референс)/B(reference); B – B(Россия)_v1/B(Russia)_v1; C – B(Россия)_v2/B(Russia)_v2; D – CRF63_02A6.
Most of the secondary structure of the Tat protein is a tangle. The consensus sequence CRF63_02A6 showed the greatest differences from the reference sequence: the absence of a helix in positions 30–33.
Tertiary structure
The tertiary structure of the consensus sequence of Tat CRF63_02A6 protein was then compared with the reference sequence (Fig. 3).

Fig. 3. Results of comparison of the tertiary structure of the consensus sequence of the Tat protein CRF63_02A6 with the reference sequence.
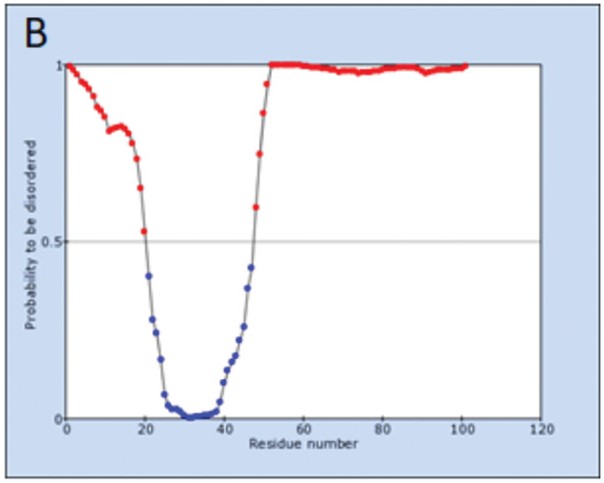
Probability profile for unstructured regions of Tat consensus sequences predicted by IsUnstruct: A – consensus sequence of HIV-1 subtype B variants circulating worldwide; B – consensus sequence of HIV-1 CRF63_02A6 variants. Spatial structure predicted by AlphaFold 2 for Tat consensus sequences: C – consensus sequence of HIV-1 subtype B variants circulating worldwide; D – consensus sequence of HIV-1 CRF63_02A6 variants. The sequence profile corresponding to unstructured regions is marked in red.
Рис. 3. Результаты сравнения третичной структуры консенсусной последовательности белка Tat CRF63_02A6 с референсной последовательностью.
Профиль вероятности для неструктурированных участков консенсусных последовательностей белка Tat, предсказанных программой IsUnstruct: A – консенсусная последовательность вариантов ВИЧ-1 субтипа В, циркулирующих в мире; B – консенсусная последовательность вариантов ВИЧ-1 CRF63_02A6. Пространственная структура, предсказанная с помощью программы AlphaFold 2, для консенсусных последовательностей белка Tat: C – консенсусная последовательность вариантов ВИЧ-1 субтипа В, циркулирующих в мире; D – консенсусная последовательность вариантов ВИЧ-1 CRF63_02A6. Красным цветом выделен профиль последовательности и участки цепи, соответствующие неструктурированным участкам; синим цветом – структурированная область. Пояснение в тексте.
The probability profiles for the unstructured regions of the Tat protein of both the reference sequence and the consensus sequence of the CRF63_02A6 variants contained only one structured region corresponding to a cysteine-rich region around 22‒48 a.a.r., which corresponds to the 2nd and 3rd domains of the Tat protein (Fig. 3). This region is highlighted in blue on the profile and on the spatial structure predicted using AlphaFold 2 (Fig. 3).
Comparison of natural polymorphism profiles of the Tat protein
When comparing the profile of natural polymorphisms of the Tat protein of HIV-1 subtype B variants circulating in Russia and CRF63_02A6 virus variants with subtype B virus variants circulating in the world, it was found that:
- 1 sequence from the group of subtype B variants circulating worldwide contained a glutamine insertion between 76 and 77 a.a.r. – 76-77insQ.
- 1 sequence CRF63_02A6 contained a histidine insertion between 80 and 81 a.a.r., 80-81insH.
- 2 sequences from the group of subtype B variants circulating worldwide contained a premature stop codon at position 87 and one sequence contained a premature stop codon at position 100.
- 1 sequence from the group of subtype B variants circulating in Russia contained a premature stop codon at position 87.
- 3 CRF63_02A6 sequences contained a premature stop codon at position 100.
However, the detected insertions and premature stop codons had no significant difference in the frequency of occurrence between the analyzed groups.
When comparing the profile of natural polymorphisms of subtype B virus variants circulating in Russia and subtype B virus variants circulating worldwide, 21 substitutions with statistically significant differences in the frequency of occurrence were identified; however, after the Bonferroni correction, only two substitutions, S68P and R78G, had a significant difference in the frequency of occurrence (Table 1).
Table 1. Substitutions in the Tat protein with a statistically significant difference in the frequency of occurrence between HIV-1 subtype B variants circulating in the world and HIV-1 subtype B variants circulating in Russia (p < 0.05)
Таблица 1. Замены в белке Tat со статистически значимой разницей в частоте встречаемости у вариантов ВИЧ-1 субтипа В, циркулирующих в мире, и у вариантов ВИЧ-1 субтипа В, циркулирующих на территории России (p < 0,05)
Domain Участок | Substitution Замена | B World Мир | B Russia Россия | p | Domain Участок | Substitution Замена | B World Мир | B Russia Россия | p |
n = 48 | n = 35 | n = 48 | n = 35 | ||||||
I | K19Q | 1 | 5 | 0.034 | VI | R78G* | 4 | 13 | 0.0013* |
II | N24A | 0 | 3 | 0.0388 | D80N | 0 | 6 | 0.0029 | |
K29Q | 5 | 0 | 0.0489 | P81Q | 1 | 6 | 0.0148 | ||
III | T40K | 16 | 20 | 0.0307 | n = 46 | n = 34 | |||
IV | Q54R | 0 | 3 | 0.0388 | K89E | 8 | 1 | 0.0432 | |
V | Q60K | 1 | 5 | 0.034 | R93K | 7 | 0 | 0.0173 | |
T64D | 0 | 5 | 0.0069 | R93S | 7 | 13 | 0.0188 | ||
S68P* | 8 | 20 | 0.0001* | D98H | 13 | 17 | 0.0471 | ||
S70P | 11 | 17 | 0.0146 | D98N | 0 | 6 | 0.0031 | ||
VI | P77A | 5 | 0 | 0.0489 | V100D | 2 | 9 | 0.0045 | |
n = 45 | n = 34 | ||||||||
P77T | 0 | 3 | 0.0388 | D101H | 7 | 14 | 0.0107 |
Note. * ‒ significant in the χ2 test with Bonferroni correction p < 0.024. Due to the presence of premature stop codons in some sequences, the number of analyzed sequences in groups changed, since amino acids (a.a.r) located after the stop codon were not taken into account in the analysis: from 1 to 87 a.a.r, the group of HIV-1 subtype B variants circulating in the world contained 48 sequences, the group of HIV-1 subtype B variants circulating in Russia – 35 sequences; from 88 to 100 a.a.r, the group of HIV-1 subtype B variants circulating in the world contained 46 sequences, the group of HIV-1 subtype B variants circulating in Russia – 34 sequences; in 101 a.a.r. the group of HIV-1 subtype B variants circulating in the world contained 45 sequences, the group of HIV-1 subtype B variants circulating in Russia – 34 sequences.
Примечание. * ‒ позиции, достоверные по критерию χ2 с поправкой Бонферрони p < 0,0024. В связи с наличием преждевременных стоп-кодонов в некоторых последовательностях менялось число (n) анализируемых последовательностей в группах, т.к. аминокислоты, находящиеся после стоп-кодона, в анализе не учитывали: с 1 по 87 АО группа вариантов ВИЧ-1 субтипа В, циркулирующих в мире, состояла из 48 последовательностей, группа вариантов субтипа В, циркулирующих на территории России, – из 35 последовательностей; с 88 АО по 100 АО группа вариантов ВИЧ-1 субтипа В, циркулирующих в мире, ‒ из 46 последовательностей, группа вариантов ВИЧ-1 субтипа В, циркулирующих на территории России, – из 34 последовательностей; в 101 АО группа вариантов ВИЧ-1 субтипа В, циркулирующих в мире, ‒ из 45 последовательностей, группа вариантов ВИЧ-1 субтипа В, циркулирующих на территории России, – из 34 последовательностей.
Comparing the natural polymorphism profile of CRF63_02A6 variants and subtype B virus variants circulating worldwide, 54 substitutions with statistically significant differences in frequency of occurrence were identified. After the Bonferroni correction, 31 substitutions had a significant difference in the frequency of occurrence (Table 2).
Table 2. Substitutions in the Tat protein with a statistically significant difference in the frequency of occurrence between HIV-1 subtype B variants circulating in the world and HIV-1 CRF63_02A6 variants (p < 0.05)
Таблица 2. Замены в белке Tat со статистически значимой разницей в частоте встречаемости у вариантов ВИЧ-1 субтипа В, циркулирующих в мире, и у вариантов CRF63_02A6 (p < 0,05)
Domain Участок | Substitution Замена | B World Мир | CRF63_02A6 | p | Domain Участок | Substitution Замена | B World Мир | CRF63_02A6 | p |
n = 48 | n = 25 | n = 48 | n = 25 | ||||||
I | E2D | 5 | 24 | 0.0000* | V | D61S | 3 | 18 | 0.0000* |
R7N | 7 | 21 | 0.0000* | S62R | 0 | 21 | 0.0000* | ||
K12N | 4 | 20 | 0.0000* | Q63E | 10 | 0 | 0.014 | ||
K19R | 11 | 0 | 0.0094 | T64N | 12 | 1 | 0.0062 | ||
A21P | 12 | 0 | 0.0062 | T64D | 0 | 22 | 0.0000* | ||
II | T23S | 0 | 17 | 0.0000* | V67A | 16 | 0 | 0.0011 | |
N24K | 11 | 1 | 0.0385 | V67D | 1 | 5 | 0.0082 | ||
K28I | 0 | 2 | 0.0469 | V67N | 0 | 19 | 0.0000* | ||
C31S | 1 | 9 | 0.0001* | S68P | 8 | 24 | 0.0000* | ||
C31V | 0 | 2 | 0.0469 | L69V | 0 | 24 | 0.0000* | ||
F32L | 10 | 0 | 0.014 | S70P | 11 | 21 | 0.0000* | ||
F32W | 1 | 25 | 0.0000* | VI | A74L | 0 | 24 | 0.0000* | |
F32Y | 7 | 0 | 0.0446 | S75P | 4 | 25 | 0.0000* | ||
V36L | 2 | 22 | 0.0000* | Q76T | 0 | 22 | 0.0000* | ||
III | I39L | 6 | 25 | 0.0000* | P77T | 0 | 24 | 0.0000* | |
I39T | 12 | 0 | 0.0062 | D80N | 0 | 22 | 0.0000* | ||
T40N | 0 | 21 | 0.0000* | P84Q | 8 | 0 | 0.0305 | ||
G42A | 10 | 0 | 0.014 | K85E | 11 | 25 | 0.0000* | ||
IV | R53G | 1 | 20 | 0.0000* | n = 46 | n = 25 | |||
Q54R | 0 | 22 | 0.0000* | K89E | 8 | 0 | 0.0269 | ||
Q54H | 0 | 3 | 0.0143 | K90E | 1 | 23 | 0.0000* | ||
R57G | 1 | 24 | 0.0000* | E92A | 0 | 22 | 0.0000* | ||
V | A58S | 7 | 0 | 0.0446 | R93S | 7 | 25 | 0.0000* | |
A58T | 9 | 23 | 0.0000* | E94K | 6 | 25 | 0.0000* | ||
P59S | 2 | 21 | 0.0000* | D98H | 13 | 1 | 0.0141 | ||
P59T | 0 | 2 | 0.0469 | P99R | 0 | 3 | 0.0164 | ||
Q60R | 0 | 2 | 0.0469 | V100C | 1 | 14 | 0.0000* | ||
D61G | 7 | 0 | 0.0446 | V100Y | 1 | 4 | 0.0297 |
Note. * ‒ significant in the χ2 test with Bonferroni correction p < 0.0009. Due to the presence of premature stop codons in some sequences, the number of analyzed sequences in groups changed, since amino acid residues (a.a.r.) located after the stop codon were not taken into account in the analysis: from 1 to 87 a.a.r, the group of HIV-1 subtype B variants circulating in the world contained 48 sequences, the group of CRF63_02A6 variants – 25 sequences; from 88 to 100 a.a.r., the group of HIV-1 subtype B variants circulating in the world contained 46 sequences, the group of CRF63_02A6 variants – 25 sequences.
Примечание. * ‒ позиции достоверные по критерию χ2 с поправкой Бонферрони p < 0,0009. В связи с наличием преждевременных стоп-кодонов в некоторых последовательностях менялось число (n) анализируемых последовательностей в группах, т.к. аминокислоты, находящиеся после стоп-кодона, в анализе не учитывали: с 1 по 87 АО группа вариантов ВИЧ-1 субтипа В, циркулирующих в мире, состояла из 48 последовательностей, группа вариантов CRF63_02A6 – из 25 последовательностей; с 88 АК по 100 АО группа вариантов ВИЧ-1 субтипа В, циркулирующих в мире, – из 46 последовательностей, группа вариантов CRF63_02A6 – из 25 последовательностей.
Discussion
One of the main characteristics of HIV-1 is its high genetic variability, which determines the extraordinary global genetic diversity of the virus [2, 20]. Polymorphisms reflect natural variation among HIV-1 genetic variants, and some of them may have functional significance [11, 12, 21]. The high genetic variability of HIV-1 is known to result from several factors that include the operation of a specific viral enzyme, reverse transcriptase and the occurrence of escape mutations in response to the host immune system [9, 22]. Studies have shown that the Tat protein is a target for the action of the cytotoxic immune response and a number of CTL epitopes in the Tat protein have been identified (https://www.hiv.lanl.gov/content/immunology/maps/ctl/Tat.html) [9]. Thus, mutations in the Tat protein may be associated with both the virus subtype and the genetic features of the host population in which the virus is circulating. The present study is aimed at investigating the features of the Tat protein in non-A6-variants of the virus characteristic of the Russian Federation.
Comparison of consensus sequences showed that all analyzed variants of the Tat protein differed from the reference sequence, with each variant containing a unique profile of substitutions.
The consensus sequence of HIV-1 subtype B variants circulating in Russia contained a Q63E substitution at position 63, which, as noted earlier, in subtype C virus variants contributed to higher transcriptional activation in human CD4 T cells [23].
The consensus sequences CRF02_AG and CRF63_02A6 contained the highest number of substitutions, with positions 32, 34, 37, 40, 54, 57, 57, 58, 61, 62, 64, 67‒70, 74‒77, 80, 90, 92‒94 containing the same amino acid substitutions relative to the reference sequence. This result is explained by the fact that CRF63_02A6 is a recombinant form of CRF02_AG and subtype A6, which corresponds to CRF02_AG in the tat gene region (https://www.hiv.lanl.gov/components/sequence/HIV/crfdb/crfs.comp).
In turn, CRF03AB is a recombinant form of sub-subtype A6 and subtype B, which corresponds to subtype B in the tat gene region (https://www.hiv.lanl.gov/components/sequence/HIV/crfdb/crfs.comp). The consensus sequence of CRF03AB at position 87 contains a stop codon, which is characteristic of some HIV-1 subtype B variants, such as the reference strain HXB2 (K03455). The shortened version of the Tat protein containing 86 a.a.r. is functional, but some functions, such as modulation of host cell cytoskeleton modification and possibly a function in ensuring optimal replication in monocyte-macrophage lineage cells, are associated with the second exon [6].
The smaller number of substitutions relative to the reference is found in the Tat protein fragment from 1 to 51 a.a.r.: the consensus of HIV-1 subtype B variants circulating in the Russian Federation contained 1 substitution, CRF02_AG – 10, CRF03_A6B – 3, CRF63_02A6 – 8. Whereas the Tat protein fragment with 52-101 a.a.r.: consensus of HIV-1 subtype B variants circulating in the Russian Federation contained 7 substitutions, CRF02_AG – 20, CRF03_A6B – 3 and a stop codon at position 87, CRF63_02A6 – 23 (Fig. 1). This is partly due to the fact that the first three domains of the Tat protein (1‒48 a.a.r.) form the minimal region required for trans-activation of viral genome transcription [6, 8]. It has also been previously noted that, in general, the region encoded by the second exon of the tat gene is less conserved than the region encoded by the first exon [10].
As a result of the comparison of consensus sequences, it was shown that the existing differences between the primary structure of CRF63_02A6 and the reference sequence, the absence of the helix element in positions 30-33 of the secondary structure of CRF63_02A6, as predicted, did not affect the spatial structure of the protein: the most structured region was located near the 2nd and 3rd domains of both CRF63_02A6 and the reference sequence (Fig. 3).
When analyzing the profile of natural polymorphisms, it was shown that S68P and R78G substitutions were significantly more frequent in HIV-1 subtype B variants circulating in Russia than in the reference group (Table 1). At the same time, the 78RGD80 motif is a ligand for certain integrins and in this regard, presumably, the R78G substitution may affect the functional properties of the Tat protein [6]. The list of substitutions with a statistically significant difference in the frequency of occurrence in the Tat protein in HIV-1 subtype B variants circulating worldwide and in CRF63_02A6 variants corresponded to the list of substitutions identified in the consensus sequence comparison. Furthermore, it was shown that the C31S substitution was significantly more frequent in CRF63_02A6 variants. It is known that the C31S substitution is functionally significant, associated with reduced neurotoxicity of the Tat protein [12, 24].
Limitation of the study. The limitation of this study is the small selection of sequences. The study of the Tat protein features of non-A6 variants of HIV-1 characteristic for the Russian Federation on large sequence samples is an interesting direction for research, which is actualized by the gradual expansion of the spread of non-A6 variants of HIV-1 on the territory of the country [4]. The study of the influence of Tat protein features of different virus variants characteristic of the Russian Federation on Tat-TAR interaction is also a promising area for possible future studies.
Conclusion
Consensus sequences of the Tat protein of non-A6 variants of HIV-1 characteristic of the Russian Federation were obtained for the first time. It was shown that different variants of the virus have characteristic features in the primary structure of the protein. The consensus sequence CRF63_02A6 contained the largest number of amino acid substitutions, while the existing features did not affect the probability profile of the location of unstructured regions of the protein. It was shown that the R78G substitution located in a functionally significant motif was significantly more frequent in subtype B virus variants circulating in Russia than in subtype B virus variants circulating worldwide. It was determined that the functionally significant C31S substitution was significantly more frequent in CRF63_02A6 variants than in variants of subtype B virus circulating in the world. Promising fields for future research were highlighted.
About the authors
Anna I. Kuznetsova
D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya
Author for correspondence.
Email: a-myznikova@list.ru
ORCID iD: 0000-0001-5299-3081
head of laboratory of T-lymphotropic viruses, PhD, leading researcher
Russian Federation, 123098, MoscowAnastasiia A. Antonova
D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya
Email: aantonova1792@gmail.com
ORCID iD: 0000-0002-9180-9846
PhD, Researcher, Laboratory of T-lymphotropic viruses
Russian Federation, 123098, MoscowAleksey V. Lebedev
D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya
Email: lebedevalesha236@gmail.com
ORCID iD: 0000-0001-6787-9345
PhD, Researcher, Laboratory of T-lymphotropic viruses
Russian Federation, 123098, MoscowEkaterina N. Ozhmegova
D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya
Email: belokopytova.01@mail.ru
ORCID iD: 0000-0002-3110-0843
PhD, Researcher, Laboratory of T-lymphotropic viruses
Russian Federation, 123098, MoscowAnastasia V. Shlykova
Central Research Institute of Epidemiology
Email: murzakova_a.v@mail.ru
ORCID iD: 0000-0002-1390-8021
Researcher
Russian Federation, Moscow, 111123Ilya A. Lapovok
Central Research Institute of Epidemiology
Email: i_lapovok@mail.ru
ORCID iD: 0000-0002-6328-1415
PhD, Senior researcher
Russian Federation, Moscow, 111123Oxana V. Galzitskaya
D.I. Ivanovsky Institute of Virology of National Research Center for Epidemiology and Microbiology named after Honorary Academician N.F. Gamaleya; Institute of Protein Research RAS; Institute of Theoretical and Experimental Biophysics RAS
Email: ogalzit@vega.protres.ru
ORCID iD: 0000-0002-3962-1520
Doctor of Physical and Mathematical Sciences, Head of the Bioinformatics Laboratory, Chief Researcher
Russian Federation, 123098, Moscow; 142290, Moscow Region, Pushchino; 142290, Moscow Region, PushchinoReferences
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup «Assessment of Pathogens Transmissible by Blood». Human Immunodeficiency Virus (HIV). Transfus. Med. Hemother. 2016; 43(3): 203–22. https://doi.org/10.1159/000445852
- Bbosa N., Kaleebu P., Ssemwanga D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS. 2019; 14(3): 153–60. https://doi.org/10.1097/COH.0000000000000534
- Kuznetsova A.I. The role of HIV-1 polymorphism in the pathogenesis of the disease. VICh-infektsiya i immunosupressii. 2023; 15(3): 26–37. https://doi.org/10.22328/2077-9828-2023-15-3-26-37 https://elibrary.ru/cwjjai (in Russian)
- Antonova A.A., Kuznetsova A.I., Ozhmegova E.N., Lebedev A.V., Kazennova E.V., Kim K.V., et al. Genetic diversity of HIV-1 at the current stage of the epidemic in the Russian federation: an increase in the prevalence of recombinant forms. VICh-infektsiya i immunosupressii. 2023; 15(3): 61–72. https://doi.org/10.22328/2077-9828-2023-15-3-61-72 https://elibrary.ru/tpwttn (in Russian)
- Kuznetsova A.I., Gromov K.B., Kireev D.E., Shlykova A.V., Lopatukhin A.E., Kazennova E.V., et al. Analysis of tat protein characteristics in human immunodeficiency virus type 1 sub-subtype A6 (Retroviridae: Orthoretrovirinae: lentivirus: human immunodeficiency Virus-1). Voprosy virusologii. 2021; 66(6): 452–63. https://doi.org/10.36233/0507-4088-83 https://elibrary.ru/cmzgyc (in Russian)
- Li L., Dahiya S., Kortagere S., Aiamkitsumrit B., Cunningham D., Pirrone V., et al. Impact of Tat genetic variation on HIV-1 disease. Adv. Virol. 2012; 2012: 123605. https://doi.org/10.1155/2012/123605
- Kuznetsova A., Kim K., Tumanov A., Munchak I., Antonova A., Lebedev A., et al. Features of Tat protein in HIV-1 sub-subtype A6 variants circulating in the Moscow Region, Russia. Viruses. 2023; 15(11): 2212. https://doi.org/10.3390/v15112212
- Ajasin D., Eugenin E.A. HIV-1 Tat: Role in bystander toxicity. Front. Cell. Infect. Microbiol. 2020: 10: 61. https://doi.org/10.3389/fcimb.2020.00061
- Kamori D., Ueno T. HIV-1 Tat and viral latency: what we can learn from naturally occurring sequence variations. Front. Microbiol. 2017; 8: 80. https://doi.org/10.3389/fmicb.2017.00080
- Spector C., Mele A.R., Wigdahl B., Nonnemacher M.R. Genetic variation and function of the HIV-1 Tat protein. Med. Microbiol. Immunol. 2019; 208(2): 131–69. https://doi.org/10.1007/s00430- 019-00583-z
- Ranga U., Shankarappa R., Siddappa N.B., Ramakrishna L., Nagendran R., Mahalingam M., et al. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J. Virol. 2004; 78(5): 2586–90. https://doi.org/10.1128/jvi.78.5.2586-2590.2004
- Ruiz A.P., Ajasin D.O., Ramasamy S., DesMarais V., Eugenin E.A., Prasad V.R. A naturally occurring polymorphism in the HIV-1 Tat basic domain inhibits uptake by bystander cells and leads to reduced neuroinflammation. Sci. Rep. 2019; 9(1): 3308. https://doi.org/10.1038/s41598-019-39531-5
- Jin H., Li D., Lin M.H., Li L., Harrich D. Tat-based therapies as an adjuvant for an HIV-1 functional cure. Viruses. 2020; 12(4): 415. https://doi.org/10.3390/v12040415
- Asamitsu K., Fujinaga K., Okamoto T. HIV Tat/P-TEFb interaction: a potential target for novel anti-HIV therapies. Molecules. 2018; 23(4): 933. https://doi.org/10.3390/molecules23040933
- Lobanov M.Y., Sokolovskiy I.V., Galzitskaya O.V. IsUnstruct: prediction of the residue status to be ordered or disordered in the protein chain by a method based on the Ising model. J. Biomol. Struct. Dyn. 2013; 31(10): 1034–43. https://doi.org/10.1080/07391102.2012.718529
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596(7873): 583–9. https://doi.org/10.1038/s41586-021-03819-2
- Shafer R.W., Rhee S.Y., Pillay D., Miller V., Sandstrom P., Schapiro J.M., et al. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007; 21(2): 215–23. https://doi.org/10.1097/QAD.0b013e328011e691
- Berezov T.T., Korovkin B.F. Biological Chemistry [Biologicheskaya khimiya]. Moscow: Meditsina; 1998. (in Russian)
- Lobanov M.Y., Pereyaslavets L.B., Likhachev I.V., Matkarimov B.T., Galzitskaya O.V. Is there an advantageous arrangement of aromatic residues in proteins? Statistical analysis of aromatic interactions in globular proteins. Comput. Struct. Biotechnol. J. 2021; 19: 5960–8. https://doi.org/10.1016/j.csbj.2021.10.036
- Tee K.K., Thomson M.M., Hemelaar J. Editorial: HIV-1 genetic diversity, volume II. Front. Microbiol. 2022; 13: 1007037. https://doi.org/10.3389/fmicb.2022.1007037
- Bobkova M.R. Genetic diversity of human immunodeficiency viruses and antiretroviral therapy. Terapevticheskii arkhiv. 2016; 88(11): 103–11. https://doi.org/10.17116/terarkh20168811103-111 https://elibrary.ru/xeaxsb (in Russian)
- Cilento M.E., Kirby K.A., Sarafianos S.G. Avoiding drug resistance in HIV reverse transcriptase. Chem. Rev. 2021; 121(6): 3271–96. https://doi.org/10.1021/acs.chemrev.0c00967
- Gotora P.T., Brown K., Martin D.R., van der Sluis R., Cloete R., Williams M.E. Impact of subtype C-specific amino acid variants on HIV-1 Tat-TAR interaction: insights from molecular modelling and dynamics. Virol. J. 2024; 21(1): 144. https://doi.org/10.1186/s12985-024-02419-6
- Mishra M., Vetrivel S., Siddappa N.B., Ranga U., Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: Significance of dicysteine C30C31 motif. Ann. Neurol. 2008; 63(3): 366–76. https://doi.org/10.1002/ana.21292
Supplementary files

