



NGS amplification panel for HBV (Hepadnaviridae: Orthohepadnavirus) sequencing
- Authors: Chanyshev M.D.1, Vlasenko N.V.1, Roev G.V.1,2, Kotov I.A.2, Glushchenko A.G.1,2, Makashova V.V.1, Khafizov K.F.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
- Moscow Institute of Physics and Technology, National Research University
- Issue: Vol 69, No 1 (2024)
- Pages: 65-75
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/16599
- DOI: https://doi.org/10.36233/0507-4088-212
- EDN: https://elibrary.ru/cilsjh
- ID: 16599

Cite item

Abstract
Introduction. Hepatitis B virus (HBV) remains a pressing global public health concern. The clinical course of the disease, particularly its tendency towards chronicity and response to therapy, is significantly influenced by the HBV genotype and specific mutations. There is an imperative need for a straightforward, highly sensitive, and dependable method for whole genome sequencing of HBV.
Objective. Development and testing of an amplification panel for HBV whole-genome sequencing.
Materials and methods. We introduce an NGS amplification panel designed for genome sequencing of HBV on the Illumina platform. A panel consisting of 54 primers, divided into 2 pools and amplifying overlapping regions of the HBV genome up to 300 bp in length, was tested on 246 HBV DNA samples.
Results. The studied samples represented a genotypic diversity of the virus, with a pronounced predominance of the genotype specific to the Moscow region: 216, 27, 2, and 1 sample were identified as genotype D, A, B, and E, respectively. Five samples contained at least one mutation associated with antiviral therapy resistance, and twenty-three samples contained at least one mutation associated with vaccine escape described in the literature.
Conclusion. The present paper describes the stages of whole-genome sequencing of HBV, provides a laboratory protocol, nucleotide sequences of the primers and an approach to the data analysis. Using a list of clinical samples as example, the reliability of the panel is shown. The HBV panel holds immense potential for utilization in scientific research, epidemiological monitoring, and advancement of personalized medicine approaches.
Keywords
Full Text
Introduction
Hepatitis B, a liver disease caused by the hepatitis B virus (HBV), is a global public health problem. The World Health Organization (WHO) estimates that in 2019, there were 296 million people diagnosed with chronic hepatitis B, and in the same year, there were about 820,000 deaths associated with HBV, mainly due to the development of cirrhosis and hepatocellular carcinoma [1]. The HBV genome is a partially double-stranded circular DNA of 3200 bp in length containing four overlapping open reading frames (pre-S1/pre-S2/S, pre-C/C, P and X). These regions encode several proteins such as surface proteins S, M and L containing the HBs antigen (HBsAg), precore/core proteins containing the HBeAg and HBcAg antigens, polymerase (P) and protein X containing the HBxAg antigen [2].
Currently, there are 10 HBV genotypes (A to J) with nucleotide sequences differing by more than 7.5%. Each genotype is subdivided into subtypes with more than 4% nucleotide differences [3]. In contrast to the long-known major genotypes A to H, two new genotypes (I and J) have been proposed relatively recently [4, 5]. The distribution of HBV genotypes varies in different geographical regions. Genotype D is known to be predominant in Eurasian countries, including Russia, while genotype A is widespread in East Africa. Thus, regional differences among HBV genotypes reflect the diversity of the virus in different geographic locations [6]. Numerous studies indicate that HBV genotypes and subtypes, genetic variants and single nucleotide mutations may be associated with different clinical features of the disease course and outcomes such as liver cirrhosis (LC) and hepatocellular carcinoma (HCC). For example, experimental evidence suggests that genotypes B and D are less associated with the chronicity of the infection process in contrast to genotypes A and C. At the same time, genotypes A and B are characterized by a higher rates of response to interferon therapy and a better prognosis in relation to LC and HCC, compared to genotypes C and D [7].
The results of studies aimed at identifying individual mutations affecting the course and outcome of hepatitis B are not insignificant. For instance, it has been identified in a number of studies that several mutations in the reverse transcriptase (RT) domain of the HBV P gene contribute to the development of resistance to antiviral therapy. For example, it has been shown that rtV173L accumulates in the viral population against lamivudine therapy and that this mutation increases the efficiency of in vitro virus replication [8]. The rtM204I/V mutation is associated with resistance to lamivudine and telbivudine; N236T and A181T/V are associated with resistance to adefovir dipivoxil; M204V + L180M and T184A/G/I/L/S, S202G and M250V build up resistance to entecavir [9, 10]. The amino acid substitutions rtL180M, rtA181T/V, rtT184G/S, rtS202G/I, rtM204I/V, rtN236T and rtM250V are characterized as mutations building up resistance to nucleotide and nucleoside analogues (NA) therapy [11]. The substitutions rtV173L, rtA181T/V/S and rtT184G/S/A, among many others have been most likely associated with resistance to NA therapy in the Chinese population [12]. Information on the presence of these mutations is taken into consideration when selecting optimal therapy. A number of S gene mutations are known to be associated with immune response evasion. For example, amino acid substitutions K141E/I/R and G145A/R are characterized by the most proven relationship with immune response evasion, which is supported by both clinical and in vitro data [13, 14]. Mutations associated with eluding the post-vaccine response, L109R, Q129R, M133L, S143L and D144E, were identified among genotype D isolates in Jordan [15]. T116N, P120S/E, I/T126A/N/I/S, Q129H/R, M133L, K141E, P142S, D144A/E and G145R/A have been described as significant mutations in relation to hepatitis B reactivation during immunosuppressive therapy [16]. The P120T, Q129H, M133I/T, F/Y134N/L substitutions are also associated with false-negative diagnostic test errors, elusion from post-vaccine response or immunoglobulin prophylaxis [17]. P120T, T126S, Q129H, G130N, S143L, D144A and G145A/R have been reported as associated with a high risk of diagnostic errors [18]. The S gene mutations E164D, I195M, P217L and P120S were identified in a population with latent HBV infection in Northern Brazil [19]. The X gene is of particular interest as numerous studies have shown that this gene is involved in carcinogenesis. Key mutations in the X gene, such as G1613A, C1653T, T1674C/G, T1753V and A1762T/G1764A, gradually accumulate during carcinogenesis and may be risk factors for HCC development. The presence of a combination of X gene mutations can be used to predict the occurrence and progression of HCC [20]. Z. Belaiba et al. analyzed the whole HBV genome before and during long-term therapy in five patients with chronic form of the disease and identified mutations in S, C and X genes that may be associated with disease progression to LC and/or HCC [2].
Thus, although many HBV studies have focused on specific regions of the virus genome, such as the S gene or the X gene, there is a need for whole genome sequencing of HBV, for which there are various approaches. A typical method is amplification of fragments 1000–1500 bp in length and Sanger sequencing [2, 21]. Probably due to the time-consuming and costly nature of Sanger sequencing, relatively small numbers of samples are usually examined in such studies. For example, Y.V. Ostankova et al. determined the complete sequence of the HBV genome in 3 samples [22]. Five patients participated in the aforementioned study by Z. Belaiba et al. [2]. Studies with a relatively large number of samples have also been published. Su-Ru Lin et al. obtained 183 complete HBV genome sequences from 1263 serum samples positive for HBsAg [23].
Next generation sequencing (NGS) allows more samples to be analyzed with less cost and time than Sanger sequencing. There are relatively few studies in which the complete HBV genome has been determined by NGS. For example, Qin-Yan Chen et al. determined HBV sequences of the whole genome in nine patients at four different time points using next-generation sequencing (HBV long-strand amplification + Celero EZ DNA-Seq) [24]. Su-Ru Lin et al. used NGS to investigate how HBV whole genome viral quasispecies evolve and diversify in response to HBeAg seroconversion and viral control in 50 patients [25]. We developed an NGS amplification panel for rapid and reliable sequencing of the entire HBV genome and tested it on 246 clinical samples.
Materials and methods
Clinical samples
In order to select optimal conditions for sample preparation, a control blood plasma sample provided by the production department of the Central Research Institute of Epidemiology of Rospotrebnadzor was used. Afterwards, the HBV amplification panel was tested on 246 clinical blood plasma samples obtained from patients observed for HBV in the clinical center of the Central Research Institute of Epidemiology of Rospotrebnadzor. Voluntary informed consent for the use of plasma samples in the study was obtained from all study participants. Patients were not selected by age, sex, ethnicity or clinical features of the disease. The current study was approved by the local ethical committee of the Central Research Institute of Epidemiology of Rospotrebnadzor, protocol 133 of March 2, 2023. DNA isolation from plasma samples was performed using the RIBO-prep reagent kit (AmpliSens, Russia). Viral load for 210 samples was determined using HBV-Monitor-FL reagent kit (AmpliSens, Russia). The average viral load was 2.81 × 104 IU/mL. The viral load for 36 samples was unknown. Information on the sex and age of the patients and viral load for each sample can be found in Table S1 in the supplementary materials.
Selection of primers
In this study, HBV genotype E from the HBVdb database was used as a reference sequence for primer selection [26]. The process of selecting suitable primers repeats the method presented in a previous publication [27]. First, a preferred target region in the genome was selected for primer binding in the range of 60–150 bp. This region was then divided into all possible fragments in the range of 16 to 30 bp, which were additionally selected based on their melting temperature (Tm) calculated using Thermo Fisher multiple primer analyzer. Only oligonucleotides with a Tm between 62 and 64 °C were selected. Adapter sequences were then added to the selected fragments and any oligonucleotides that formed homodimers were removed. To evaluate potential primer interactions, pairwise Gibbs free energy changes between each oligonucleotide and primers previously included in the pool were calculated. The PrimerDimer program was used to calculate the Gibbs free energy change [28]. As a result, one oligonucleotide with the best total score of the pairwise Gibbs free energy change was added to the pool. This process was repeated many times, moving from the 5’-end to the 3’-end of the genome, until the entire viral genome was covered with primers. During the panel testing process, some primers were replaced with manually selected primers. As a result, 54 primers were selected for amplification of the viral genome divided into 20 regions.
HBV Sequencing
To amplify the entire HBV genome, we used a panel of primers containing Illumina adapter sequences at the ends. Multiplex polymerase chain reaction (PCR) amplification was performed in two separate reactions containing 10 μl of matrix DNA, 10 μl of PCR-mix-2-blue (AmpliSens, Russia), 1.4 μl of 4.4 mM dNTP (AmpliSens, Russia), primers (the final concentration of each primer in the reaction mixture and pool number are listed in Table S2 in the supplementary materials), and sterile nuclease-free water in a final volume of 25 μl. As a result, at an average viral load of 1.12 × 105 IU/mL, 2.24 × 103 copies were present in the PCR mixture. The amplification protocol was (1) denaturation at 95 °C for 3 min; (2) 16 cycles: 95 °C – 30 s, 55 °C – 30 s, 72 °C – 20 s; (3) final elongation at 72 °C for 3 min. The mixed PCR products were purified using AMPure XP magnetic particles (Beckman Coulter, USA) at a 1 : 1 ratio, eluting in 15 μl of 0.1x TE. PCR indexing was performed in a 25 μl reaction volume containing 10 μl of PCR-mix-2-blue (AmpliSens, Russia), 1.4 μl of 4.4 mm dNTP (AmpliSens, Russia), 5 μl of purified PCR products, sterile nuclease-free water, and Nextera index adapters, the final concentration of each primer was 200 nM. Amplification profile: (1) denaturation at 95 °C for 1 min; (2) 25 cycles: 95 °C – 20 s, 55 °C – 30 s, 72 °C – 20 s; (3) final elongation at 72 °C for 3 min. PCR products were visualized by electrophoresis in a 1.7% agarose gel stained with ethidium bromide. Mixed PCR products were purified using AMPure XP magnetic particles (Beckman Coulter USA) at a 1 : 1 ratio. The concentration of the purified library was measured using the Qubit dsDNA HS Assay Kit on a Qubit 4.0 fluorimeter (Invitrogen, Waltham, MA, USA). High-throughput sequencing was performed on the Illumina MiSeq platform using the MiSeq v2 reagent kit (300 and 500 cycles) or the MiSeq v3 reagent kit (600 cycles).
Bioinformatics analysis
The resulting reads were screened by sequencing quality control using the FastQC program. Adapter sequences were removed from the reads using Trimmomatic software, and primer sequences were removed using Cutadapt software. Next, the processed reads were aligned to the reference sequence using Bowtie2 software. Finally, the sequences were imported from BAM format files using the iVar program.
HBV genome assembly program
Together with the primer panel, we present a program for processing the data obtained with our method. The program is based on several key ideas: 1) only reads containing known primers at the ends are considered; 2) reads should also be similar to known HBV genotypes; 3) the quality of forward reads is usually better than the quality of reverse reads, so when overlapping paired-end reads, forward reads are considered; 4) the quality of reads usually decreases towards the end, so when overlapping amplicons are used, the amplicon with the higher number is favored. Despite the large number of objections, this scheme allows for quick and easy generation of HBV genomes from NGS reads. Moreover, in many cases the program produces the correct result, whereas standard bioinformatics processing leads to errors, mainly related to mismatches between the true and reference sequences. Using raw NGS data, the program assembles HBV genomes and performs analysis of read coverage and heterogeneity. It can be run from any Python development environment or from the command line in any operating system. The program and a detailed description of it are available at https://github.com/ChanyshevMD/HBV_seq.
Panel validation
To validate the HBV panel, we sequenced three samples of different genotypes (sample 1: A2, sample 2: D3, sample 3: B2) using an alternative approach. HBV DNA was amplified using three primer pairs AF: 5’-AAGAACTCCCTCGCCTC-3’, AR: 5’-GATGATGGGATGGGAATACARGTG-3’, BF: 5’-GGTATGTTGCCCGTTTGTCC-3’, BR: 5’-GCWAGGAGTTCCGCAGTATGG-3’, CF: 5’-TGCCAAGTGTTTGCTGACGC- 3’, CR: 5’-TGAGATCTTCTGCGACGCGG-3’.
The amplicon lengths for each primer pair AF/AR, BF/BR, and CF/CR were 1460, 829, and 1257 bp, respectively (isolate P2-121214, GenBank: AB981583.1). The AF/AR and BF/BR primer pairs were used in the study by Hebeler-Barbosa et al. [29]. The PCR mixture contained 10 μl of matrix DNA, 10 μl of PCR-mix-2-blue (AmpliSens, Russia), 1.4 μl of 4.4 mm dNTP (AmpliSens, Russia), primers with a final concentration of 200 nM and sterile nuclease-free water in a final volume of 25 μl. Amplification protocol: (1) denaturation at 95°C for 3 min; (2) 16 cycles: 95 °C – 30 s, 55 °C – 30 s, 72 °C – 90 s; (3) final elongation at 72 °C for 3 min. The length of PCR products obtained was verified by electrophoresis in 1% agarose gel. Purified PCR products were quantified using a Qubit 2.0 fluorimeter (Invitrogen) and a DNA High Sensitivity Assay kit (ThermoFisher). For each sample, the three PCR products (A, B, and C) were diluted to 0.2 ng/μL and mixed at equimolar concentrations. Library preparation was performed using the Nextera XT DNA kit (Illumina) according to the manufacturer’s instructions, changing the concentration of the injected DNA to 0.5 ng (amplicons A, B, and C for each sample). The indexed Nextera library was further processed as the standard HBV panel library after indexing. Quality control of the reads was performed using FastQC. Removal of adapter sequences from the reads was performed using the Trimmomatic program, and removal of primer sequences using the Cutadapt program. Next, the processed reads were aligned to the reference sequence using bwa-mem software. In the last step, sequences were imported from bam format files using the iVar program. Furthermore, the nucleotide sequences of PCR products were determined by Sanger sequencing, which was performed in a specialized department of the Central Research Institute of Epidemiology using 3500xL Genetic Analyzer (Applied Biosystems, USA) and BigDye Terminator v1.1 Cycle Sequencing Kit (ThermoFisher, USA). Sequencing results using our HBV panel, Nextera and Sanger matched almost completely. Files of raw NGS and Sanger reads for all three samples can be downloaded at https://github.com/ChanyshevMD/HBV_seq.
Building a phylogenetic tree
The nucleotide sequences obtained in this study and 199 HBV reference genomes (supplementary materials) from the HBVdb database [26] representing genotypes A, B, D, and E were used to construct the phylogenetic tree. Multiple sequence alignment was performed using MAFFT v7.490 [30]. The tree was constructed in MEGA v11.0.11 [31] using the neighbor joining algorithm and rooted using the midpoint method. Visualization was done using the ete3 library [32]. Four samples (Sample_019, Sample_082, Sample_089, Sample_133) were not genotyped due to low coverage and therefore not used in finding the location on the phylogenetic tree.
Results
Sequencing of clinical samples
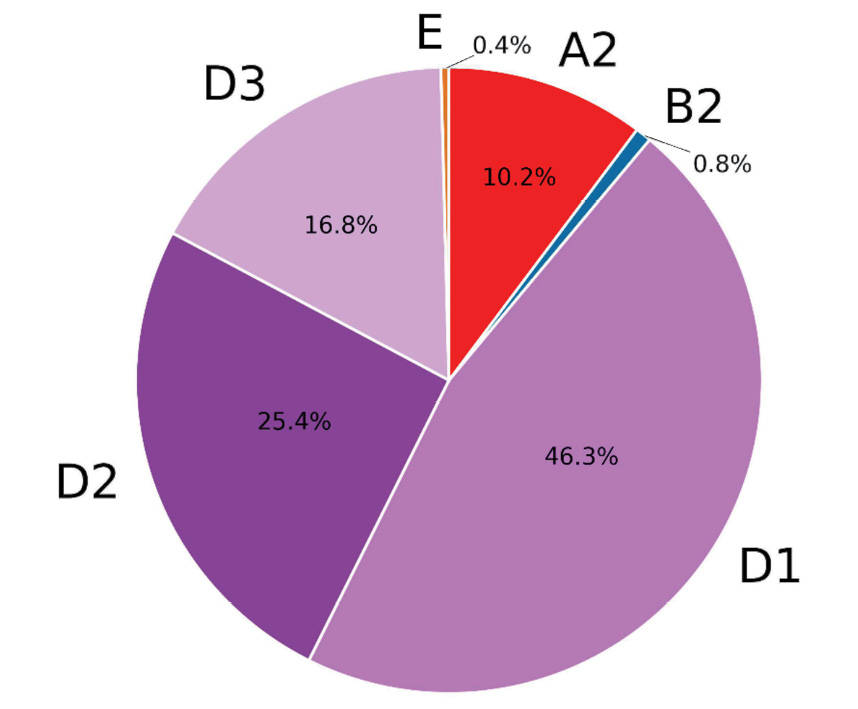
Within the framework of the present study, 246 clinical samples from patients, observed for hepatitis B in Moscow medical institutions, were sequenced and analyzed. The amplicon read depth is presented in Figure 1. Consensus sequences were assembled using the program described above and validated by comparison with consensus sequences obtained by standard bioinformatics methods. The sequences obtained are presented in the HBV_samples.fa file in the supplementary materials. The average percentage of coverage of the HBV genome was 96.3%. At the same time, 126 samples were read at 100% (Figure 2). It seems that the low coverage percentage of some samples may be due to insufficient quantity and quality of HBV DNA. Genotypes were determined by similarity to reference genomes; in ambiguous cases, a phylogenetic tree was constructed (Figure 3). The majority of samples were classified as genotype D and accounted for 88%. Subtype D1 was identified in 113 cases, D2 was represented by 62 samples, and subtype D3 was identified in 41 samples examined. 11% of the samples represented genotype A, exclusively subtype A2, two studied samples were determined as genotype B2, and in one case genotype E was determined. The phylogenetic tree is presented in Figure 4 which has a notable diversity of genetic variants.

Fig. 1. Log10 transformed read counts per amplicon norma by total reads per sample.
HBV genome was divided into 20 amplicons (HBV-1–HBV-20).
Рис. 1. Log10 глубины прочтения для каждого ампликона, нормированной на общее количество прочтений на образец.
Весь геном ВГВ был поделен на 20 ампликонов (HBV-1–HBV-20).

Fig. 2. Coverage of 246 samples.
The genome region was considered as covered at a minimum depth of 10 reads. Samples are sorted by coverage. The color designation of HBV genotypes corresponds to that from S. Velkov et al. [6].
Рис. 2. Покрытие 246 образцов.
Область генома считали прочитанной при глубине не менее 10 прочтений. Образцы отсортированы по проценту покрытия. Генотипы выделены цветами в соответствии со статьей S. Velkov и соавт. [6].

Fig. 3. Occurrence of various HBV genotypes in the Moscow region of Russia.
The color designation of HBV genotypes corresponds to that from [6].
Рис. 3. Встречаемость различных генотипов ВГВ в Московской области России.
Генотипы выделены цветами в соответствии со статьей S. Velkov и др. [6].

Fig. 4. Phylogenetic tree for sequenced HBV samples.
The tree was constructed in MEGA v11.0.11 using the neighbor joining algorithm and rooted using the midpoint method. Visualization was made using the ete3 library. Genotypes A, B, D, E are marked in red, blue, violet, and orange, respectively. References and sequences from this study are highlighted in black and yellow, respectively.
Рис. 4. Филогенетическое дерево секвенированных образцов ВГВ.
Дерево было построено в MEGA v11.0.11 с использованием алгоритма присоединения соседей и укоренено при помощи метода средней точки. Визуализация выполнена с использованием библиотеки ete3. Генотипы A, B, D, E отмечены соответственно красным, синим, фиолетовым и оранжевым цветами. Референсные последовательности помечены черным цветом, а секвенированные в данной работе – желтым.
Subsequent analysis of sequences included the identification and estimation of the prevalence of HBV mutations of significance with respect to therapy and infection prevention. Such mutations include substitutions in the RT-domain of the P gene: L80I/V, I169T, V173L/M, L180M, A181S, T184I/L/S, S202G, M204V/I, N236T, M250V associated with resistance to antiviral therapy, as well as substitutions in the S gene such as L109R, T116N, P120S/T/E, T126A/N/I/S, Q129H/R, G130N, M133L/I/T, F/Y134N/L, K141E/I/R, P142S, S/T143L, D144A/E, G145A/R, E164D, I195M, associated with immune response evasion. Five samples contained at least one mutation associated with resistance to antiviral therapy, and in twenty-three samples at least one mutation associated with immune evasion was found. In addition, eight samples contained at least one amino acid substitution at sites of mutations associated with immune evasion. The mutations are presented in Figure 5. An equally important issue in scientific activity is the search for and in-depth study of virus mutations that have a significant impact on the prognosis of the underlying disease and the rate of development of liver pathology. According to the results of numerous studies aimed at forming an idea of specific markers characterizing potentially unfavorable course of hepatitis B virus infection, the main attention is paid to X, precore and core HBV genes. Thus, W28* and G29D substitutions in the precore region were detected in 69% and 33% of samples, respectively. Amino acid substitutions F24Y, E64D, E77Q, A80I/T/V, L116I and cE180A in the core region were found in 10.5, 22, 10, 38, 44 and 0.4%, respectively. In the HBV X-gene, nucleotide substitutions G1613A, C1653T, T1674S, T1753V, A1762T, G1764A and T1768A were found in 9, 11, 11, 26, 26, 33 and 2%, respectively. These substitutions, according to the literature [20, 33], are markers indicating the likelihood of an unfavorable course of HBV and a high rate of HCC development. The prevalence of these HBV mutations is relatively high in the studied sample of patients in the Moscow region. This fact, undoubtedly, indicates the necessity for further monitoring of the virus circulating in the region in order to form prognostic models and optimize the provision of qualified medical care.

Fig. 5. The detection rates of mutations in RT domain of P gene associated with resistance to antiviral therapy and mutations in S gene associated with evasion of vaccine-induced response.
Gray, white, yellow, and red boxes represent respectively the undetermined regions, wild type, substitutions not described in the literature, and substitutions described in the literature data as associated with resistance to antiviral therapy and vaccine escape mutations. Total 33 samples containing at least one difference from the wild type are presented.
Рис. 5. Встречаемость мутаций в RT-домене гена P, связанных с устойчивостью к противовирусной терапии, и мутаций в гене S, связанных с ускользанием от поствакцинального ответа.
Серые поля представляют собой неопределенные области, белые – дикий тип, желтые – замены, не описанные в литературе, красные – мутации, описанные в литературе как мутации устойчивости к противовирусной терапии и мутации, ускользания от иммунитета. Представлено всего 33 образца, содержащих хотя бы одно отличие от дикого типа.
Discussion
The data obtained in the present study on the genotypic diversity of HBV present in the Moscow region are consistent with the few published results of colleagues. Thus, in the study conducted by V.V. Klushkina et al., based on the results of sequencing of the S-gene region in 63 serum samples from the Moscow region, genotype D was detected in 84% of the studied samples (52/63), genotype A – in 14% (9/63) and genotype C – in 2% (1/63). Furthermore, genotype D was represented by four subtypes: D1, D2, D3 and D4; genotype A consisted only of subtype A2 and genotype C only of subtype C1 [34]. According to the study by V.A. Manuilov et al., the distribution of HBV genotypes in samples of urban population of the former western USSR (n = 179) represents: D – 82%, A – 17%, C – 1%. Genotype D was represented by subtypes D1, D2, and D3, genotype A by subtype A2, and C by subtype C2 [35]. Furthermore, our data are consistent with a review article on the prevalence of HBV genotypes worldwide [6]. Comparison of the prevalence of mutations associated with resistance to antiviral therapy or evasion of vaccine-induced response is difficult due to differences in therapeutic approaches in different countries. For example, in Russia, almost the entire population is vaccinated against HBV, but antiviral therapy with nucleoside or nucleotide analogs is hardly ever used. The aforementioned article by V.A. Manuilov et al. presents the nucleotide sequences of HBV from the urban population of the former western USSR. 2 out of 179 samples contain at least one mutation associated with resistance to antiviral therapy (rtL173V KX925288 and rtS202G AY603465), and 14 samples contain at least one mutation associated with immune response evasion. Furthermore, eight samples contain at least one amino acid substitution at the sites of mutations arising in viral genomes in response to vaccination. The mutations are plotted in Figure S1 provided in the supplementary materials. Thus, the occurrence of mutations in the samples from the study conducted by V.A. Manuilov et al. is consistent with the results obtained in this study: several antiviral therapy resistance mutations, and about twenty mutations associated with immune response evasion, among two hundred samples.
Mutations of X, precore and core regions of HBV, characterized in scientific literature as markers of unfavorable course and outcomes of the disease, are also addressed in the present study, due to the importance of studying functional substitutions in these regions and the relatively high prevalence of these substitutions. This kind of research aimed at a close study of individual HBV mutations and their effect on the human body undoubtedly requires a rapid as well as an accurate method of determining the nucleotide sequence of the circulating pathogen. The results obtained in the present study indicate the possibility of using the amplification panel to perform HBV whole genome sequencing using NGS and subsequent analysis. The applications of the data obtained cover various areas of scientific interest, both from the side of classical epidemiology and from the point of view of developing a personalized approach to patient management. On the one hand, the use of whole genome sequencing methods will allow to use the obtained data in routine scientific practice during epidemiological studies to assess and analyze the prevalence of genotypes and subtypes of the virus in the territory of the Russian Federation, in the formation of prognostic epidemiological models of the development of the epidemic process, in epidemiological investigations, etc. On the other hand, this approach will make it possible to solve narrow scientific tasks of identifying nucleotide substitutions both actively studied in the world and detected for the first time.
Conclusion
This study thoroughly describes the steps of HBV whole genome sequencing, the laboratory protocol, nucleotide sequences of the primers used, and the approach to analyzing the data obtained. The validity of the applied panel in particular, and of the applied method in general, is demonstrated using clinical samples. The methodology demonstrated in this study has great potential for various purposes. This approach can be widely used in research projects to advance knowledge of HBV and to identify complex relationships between genetic variants of the pathogen and clinical outcomes of the disease. Furthermore, HBV whole genome sequencing can assist with the optimization of the epidemiological surveillance system, allowing effective monitoring of the dynamics of the pathogen. It is noteworthy that understanding the dynamics of the viral genome mutation process and the fixation of certain variants in the population can ultimately ensure the implementation of personalized therapy programs, allowing informed decisions to be made, taking into account the specific genetic profiles of HBV in patients.
About the authors
Mikhail D. Chanyshev
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Author for correspondence.
Email: chanishq@gmail.com
ORCID iD: 0000-0002-6943-2915
PhD (Biol.), Researcher of the Laboratory for genomic research, Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, MoscowNatalia V. Vlasenko
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Email: vlasenko@cmd.su
ORCID iD: 0000-0002-2388-1483
Researcher of the Laboratory of Viral Hepatitis, Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, MoscowGerman V. Roev
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing; Moscow Institute of Physics and Technology, National Research University
Email: roevherman@gmail.com
ORCID iD: 0000-0002-2353-5222
биоинформатик Лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Russian Federation, 111123, Moscow; 115184, DolgoprudnyIvan A. Kotov
Moscow Institute of Physics and Technology, National Research University
Email: ivan.kotov@phystech.edu
ORCID iD: 0000-0003-2416-5689
PhD student, Phystech School of biological and medical physics of MIPT
Russian Federation, 115184, DolgoprudnyAlbina G. Glushchenko
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing; Moscow Institute of Physics and Technology, National Research University
Email: albinagluschenko@gmail.com
ORCID iD: 0009-0002-8851-8703
laboratory assistant of the Laboratory for genomic research, Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, Moscow; 115184, DolgoprudnyVera V. Makashova
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Email: veramakashova@yandex.ru
ORCID iD: 0000-0002-0982-3527
Doctor of Medical Sciences, Professor, Leading Researcher of Clinical Department of Infectious Pathology Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, MoscowKamil F. Khafizov
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Email: kkhafizov@gmail.com
ORCID iD: 0000-0001-5524-0296
PhD (Biol.), Head of the Laboratory for genomic research, Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, MoscowVasily G. Akimkin
Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
Academician of the Russian Academy of Sciences, Doctor of Medical Sciences, Professor, Director of Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Russian Federation, 111123, MoscowReferences
- WHO. Hepatitis B; 2022. Available at: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b
- Belaiba Z., Ayouni K., Gdoura M., Kammoun Rebai W., Touzi H., Sadraoui A., et al. Whole genome analysis of hepatitis B virus before and during long-term therapy in chronic infected patients: Molecular characterization, impact on treatment and liver disease progression. Front. Microbiol. 2022; 13: 1020147. https://doi.org/10.3389/fmicb.2022.1020147
- Kramvis A. Genotypes and genetic variability of hepatitis B virus. Intervirology. 2014; 57(3-4): 141–50. https://doi.org/10.1159/000360947
- Tran T.T., Trinh T.N., Abe K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008; 82(11): 5657–63. https://doi.org/10.1128/JVI.02556-07
- Tatematsu K., Tanaka Y., Kurbanov F., Sugauchi F., Mano S., Maeshiro T., et al. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009; 83(20): 10538–47. https://doi.org/10.1128/JVI.00462-09
- Velkov S., Ott J.J., Protzer U., Michler T. The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes (Basel). 2018; 9(10): 495. https://doi.org/10.3390/genes9100495
- Sunbul M. Hepatitis B virus genotypes: global distribution and clinical importance. World J. Gastroenterol. 2014; 20(18): 5427–34. https://doi.org/10.3748/wjg.v20.i18.5427
- Delaney W.E. 4th., Yang H., Westland C.E., Das K., Arnold E., Gibbs C.S., et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003; 77(21): 11833–41. https://doi.org/10.1128/jvi.77.21.11833-11841.2003
- Zhang X., Chen X., Wei M., Zhang C., Xu T., Liu L., et al. Potential resistant mutations within HBV reverse transcriptase sequences in nucleos(t)ide analogues-experienced patients with hepatitis B virus infection. Sci. Rep. 2019; 9(1): 8078. https://doi.org/10.1038/s41598-019-44604-6
- Vincenti D., Piselli P., Solmone M., D’Offizi G., Capobianchi M.R., Menzo S. Evolutionary trends of resistance mutational patterns of HBV reverse transcriptase over years (2002–2012) of different treatment regimens: The legacy of lamivudine/adefovir combination treatment. Antiviral. Res. 2017; 143: 62–8. https://doi.org/10.1016/j.antiviral.2017.03.008
- Araujo N.M., Teles S.A., Spitz N. Comprehensive analysis of clinically significant hepatitis B virus mutations in relation to genotype, subgenotype and geographic region. Front. Microbiol. 2020; 11: 616023. https://doi.org/10.3389/fmicb.2020.616023
- Liu Y., Wang C., Zhong Y., Li X., Dai J., Ren X., et al. Genotypic resistance profile of hepatitis B virus (HBV) in a large cohort of nucleos(t)ide analogue-experienced Chinese patients with chronic HBV infection. J. Viral. Hepat. 2011; 18(4): e29–39. https://doi.org/10.1111/j.1365-2893.2010.01360.x
- Carman W.F. The clinical significance of surface antigen variants of hepatitis B virus. J. Viral. Hepat. 1997; 4(Suppl. 1): 11–20. https://doi.org/10.1111/j.1365-2893.1997.tb00155.x
- Mokaya J., Vasylyeva T.I., Barnes E., Ansari M.A., Pybus O.G., Matthews P.C. Global prevalence and phylogeny of hepatitis B virus (HBV) drug and vaccine resistance mutations. J. Viral. Hepat. 2021; 28(8): 1110–20. https://doi.org/10.1111/jvh.13525
- Ababneh N.A., Sallam M., Kaddomi D., Attili A.M., Bsisu I., Khamees N., et al. Patterns of hepatitis B virus S gene escape mutants and reverse transcriptase mutations among genotype D isolates in Jordan. PeerJ. 2019; 7: e6583. https://doi.org/10.7717/peerj.6583
- Lazarevic I., Banko A., Miljanovic D., Cupic M. Immune-escape hepatitis B virus mutations associated with viral reactivation upon immunosuppression. Viruses. 2019; 11(9): 778. https://doi.org/10.3390/v11090778
- Avellón A., Echevarria J.M. Frequency of hepatitis B virus ‘a’ determinant variants in unselected Spanish chronic carriers. J. Med. Virol. 2006; 78(1): 24–36. https://doi.org/10.1002/jmv.20516
- Ma Q., Wang Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J. Med. Virol. 2012; 84(2): 198–206. https://doi.org/10.1002/jmv.23183
- Araújo S.D.R., Malheiros A.P., Sarmento V.P., Nunes H.M., Freitas P.E.B. Molecular investigation of occult hepatitis B virus infection in a reference center in Northern Brazil. Braz. J. Infect. Dis. 2022; 26(3): 102367. https://doi.org/10.1016/j.bjid.2022.102367
- Zhou X., Liu D., Li Z., Zhao J., Cai S., Cao G. The mechanism of hepatitis B virus X gene in promoting hepatocellular carcinoma. J. Cancer Sci. Clin. Ther. 2022; 6(2): 222–33. https://doi.org/10.26502/jcsct.5079158
- Abdou Chekaraou M., Brichler S., Mansour W., Le Gal F., Garba A., Dény P., et al. A novel hepatitis B virus (HBV) subgenotype D (D8) strain, resulting from recombination between genotypes D and E, is circulating in Niger along with HBV/E strains. J. Gen. Virol. 2010; 91(Pt. 6): 1609–20. https://doi.org/10.1099/vir.0.018127-0
- Ostankova Yu.V., Semenov A.V., Zueva E.B., Totolyan A.A. The first cases of hepatitis B Virus Subgenotype D4 detection in patients with chronic, acute, and occult hepatitis B in the Russian Federation. Molekulyarnaya genetika, mikrobiologiya i virusologiya. 2020; 35(4): 221–8. https://doi.org/10.3103/S0891416820040072 https://elibrary.ru/bbamjw
- Liu H., Shen L., Zhang S., Wang F., Zhang G., Yin Z., et al. Complete genome analysis of hepatitis B virus in Qinghai-Tibet plateau: the geographical distribution, genetic diversity, and co-existence of HBsAg and anti-HBs antibodies. Virol. J. 2020; 17(1): 75. https://doi.org/10.1186/s12985-020-01350-w
- Chen Q.Y., Jia H.H., Wang X.Y., Shi Y.L., Zhang L.J., Hu L.P., et al. Analysis of entire hepatitis B virus genomes reveals reversion of mutations to wild type in natural infection, a 15 year follow-up study. Infect. Genet. Evol. 2022; 97: 105184. https://doi.org/10.1016/j.meegid.2021.105184
- Lin S.R., Yang T.Y., Peng C.Y., Lin Y.Y., Dai C.Y., Wang H.Y., et al. Whole genome deep sequencing analysis of viral quasispecies diversity and evolution in HBeAg seroconverters. JHEP Rep. 2021; 3(3): 100254. https://doi.org/10.1016/j.jhepr.2021.100254
- HBVdb. Available at: https://hbvdb.lyon.inserm.fr/HBVdb/HBVdbIndex
- Kotov I., Saenko V., Borisova N., Kolesnikov A., Kondrasheva L., Tivanova E., et al. Effective approaches to study the genetic variability of SARS-CoV-2. Viruses. 2022; 14(9): 1855. https://doi.org/10.3390/v14091855
- Johnston A.D., Lu J., Ru K.L., Korbie D., Trau M. PrimerROC: accurate condition-independent dimer prediction using ROC analysis. Sci. Rep. 2019; 9(1): 209. https://doi.org/10.1038/s41598-018-36612-9
- Hebeler-Barbosa F., Wolf I.R., Valente G.T., Mello F.C.D.A., Lampe E., Pardini M.I.M.C., et al. A new method for next-generation sequencing of the full hepatitis B virus genome from a clinical specimen: impact for virus genotyping. Microorganisms. 2020; 8(9): 1391. https://doi.org/10.3390/microorganisms8091391
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30(4): 772–80. https://doi.org/10.1093/molbev/mst010
- Tamura K., Stecher G., Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021; 38(7): 3022–7. https://doi.org/10.1093/molbev/msab120
- Huerta-Cepas J., Serra F., Bork P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016; 33(6): 1635–8. https://doi.org/10.1093/molbev/msw046
- Al-Qahtani A.A., Al-Anazi M.R., Nazir N., Abdo A.A., Sanai F.M., Al-Hamoudi W.K., et al. The correlation between hepatitis B virus precore/core mutations and the progression of severe liver disease. Front. Cell Infect. Microbiol. 2018; 8: 355. https://doi.org/10.3389/fcimb.2018.00355
- Klushkina V.V., Kyuregyan K.K., Kozhanova T.V., Popova O.E., Dubrovina P.G., Isaeva O.V., et al. Impact of universal hepatitis В vaccination on prevalence, infection-associated morbidity and mortality, and circulation of immune escape variants in Russia. PLoS One. 2016; 11(6): e0157161. https://doi.org/10.1371/journal.pone.0157161
- Manuylov V., Chulanov V., Bezuglova L., Chub E., Karlsen A., Kyuregyan K., et al. Genetic diversity and possible origins of the hepatitis B virus in Siberian natives. Viruses. 2022; 14(11): 2465. https://doi.org/10.3390/v14112465
Supplementary files

