



Амплификационная панель NGS для секвенирования ДНК вируса гепатита В (Hepadnaviridae: Orthohepadnavirus)
- Авторы: Чанышев М.Д.1, Власенко Н.В.1, Роев Г.В.1,2, Котов И.А.2, Глущенко А.Г.1,2, Макашова В.В.1, Хафизов К.Ф.1, Акимкин В.Г.1
-
Учреждения:
- ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
- Московский физико-технический институт
- Выпуск: Том 69, № 1 (2024)
- Страницы: 65-75
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://virusjour.crie.ru/jour/article/view/16599
- DOI: https://doi.org/10.36233/0507-4088-212
- EDN: https://elibrary.ru/cilsjh
- ID: 16599

Цитировать

Аннотация
Введение. Гепатит В является актуальной проблемой общественного здравоохранения во всем мире. На клиническое течение заболевания, особенно на его склонность к хронизации инфекции и развитию устойчивости к терапии, значительное влияние оказывают генотип и специфические мутации вируса гепатита В (ВГВ). С учетом сохраняющейся важности эпидемиологического контроля и профилактики заболевания, существует необходимость в простом, высокочувствительном и надежном методе секвенирования полного генома ВГВ.
Цель работы. Создание и апробация амплификационной панели для полногеномного секвенирования ВГВ.
Материалы и методы. В настоящей работе мы представляем амплификационную панель NGS, предназначенную для секвенирования генома ВГВ на платформе Illumina. Панель, состоящая из 54 праймеров, разделенных на 2 пула и амплифицирующих перекрывающиеся участки генома ВГВ длиной до 300 п.н., была апробирована на 246 образцах ДНК ВГВ, выделенных из крови.
Результаты. Исследуемая выборка представляла собой широкое генотипическое разнообразие вируса, с выраженным преобладанием генотипа, характерного для Московского региона: 216 образцов были определены как генотип D, 27 – как генотип A, 2 – генотип B и 1 – генотип E. Пять образцов содержали по меньшей мере одну мутацию, связанную с устойчивостью к противовирусной терапии, в 23 образцах была найдена по меньшей мере одна мутация, связанная с ускользанием от поствакцинального ответа.
Заключение. В работе детально изложены этапы проведения полногеномного секвенирования ВГВ, приведены лабораторный протокол, нуклеотидные последовательности используемых праймеров и подход к анализу полученных данных. На примере выборки клинических образцов показана состоятельность применяемой панели. Панель для секвенирования ВГВ обладает большим потенциалом для использования в научных исследованиях, эпидемиологическом мониторинге и развитии методов персонализированной медицины.
Ключевые слова
Полный текст
Введение
Гепатит В, заболевание печени, вызываемое вирусом гепатита В (ВГВ), является глобальной проблемой общественного здравоохранения во всем мире. По оценкам Всемирной организации здравоохранения (ВОЗ), в 2019 г. насчитывалось 296 млн больных с диагнозом «хронический гепатит В» и в том же году было зарегистрировано около 820 тыс. смертельных исходов, связанных с гепатитом В, в основном вследствие развития цирроза печени (ЦП) и гепатоцеллюлярной карциномы (ГЦК) [1]. Геном ВГВ представляет собой частично двухцепочечную кольцевую ДНК длиной 3200 п.н., содержащую четыре перекрывающиеся открытые рамки считывания (pre-S1/pre-S2/S, pre-C/C, P и X). Эти участки кодируют несколько белков, таких как поверхностные белки S, M и L, содержащие антиген HBs (HBsAg), белки precore/core, содержащие антигены HBeAg и HBcAg, полимеразу (P), и белок X, содержащий антиген HBxAg [2].
В настоящее время выделяют 10 генотипов ВГВ (от A до J), нуклеотидная последовательность которых различается более чем на 7,5%. Каждый генотип подразделяется на подтипы, имеющие более 4% нуклеотидных различий [3]. В отличие от известных уже долгое время основных генотипов от A до H, два новых генотипа (I и J) были предложены относительно недавно [4, 5]. Распределение генотипов ВГВ варьируется в разных географических регионах. Известно, что генотип D преобладает в странах Евразии, включая Россию, в то время как генотип А широко распространен в Восточной Африке. Таким образом, региональные различия среди генотипов ВГВ отражают многообразие вируса в разных географических точках [6]. Многочисленные исследования показывают, что генотипы и подтипы ВГВ, генетические варианты и однонуклеотидные мутации вируса могут быть связаны с различными клиническими особенностями течения заболевания и такими исходами, как ЦП и ГЦК. Так, экспериментальные данные свидетельствуют о том, что генотипы B и D в меньшей степени связаны с хронизацией инфекционного процесса, в отличие от генотипов A и C. В то же время генотипы A и B характеризуются высоким ответом к интерферонотерапии и лучшим прогнозом в отношение ЦП и ГЦК по сравнению с генотипами C и D [7].
Немаловажными являются результаты исследований, направленных на выявление отдельных мутаций, влияющих на течение и исход гепатита B. Так, ряд мутаций в домене обратной транскриптазы (RT) гена P ВГВ был определен в исследованиях качестве способствующих развитию резистентности к противовирусной терапии. Например, было показано, что rtV173L накапливается в вирусной популяции на фоне терапии ламивудином, а также что эта мутация повышает эффективность репликации вируса in vitro [8]. Мутация rtM204I/V ассоциирована с устойчивостью к ламивудину и телбивудину; N236T и A181T/V связаны с устойчивостью к препарату адефовир дипивоксил; M204V + L180M и T184A/G/I/L/S, S202G и M250V приводят к резистентность к энтекавиру [9, 10]. Аминокислотные замены rtL180M, rtA181T/V, rtT184G/S, rtS202G/I, rtM204I/V, rtN236T и rtM250V характеризуются как мутации, приводящие к резистентности к терапии аналогами нуклеотидов и нуклеозидов (АН) [11]. Замены rtV173L, rtA181T/V/S и rtT184G/S/A среди многих других были вероятнее всего связаны с резистентностью к терапии АН в китайской популяции [12]. Информация о наличии этих мутаций принимается во внимание при выборе оптимальной терапии. Известен ряд мутаций гена S, связанных с ускользанием от иммунного ответа. Например, аминокислотные замены K141E/I/R и G145A/R характеризуются наиболее доказанной взаимосвязью с ускользанием от иммунитета, что подтверждается как с помощью клинических данных, так и in vitro [13, 14]. Мутации L109R, Q129R, M133L, S143L и D144E, ассоциированные с ускользанием от поствакцинального ответа, были выявлены среди изолятов генотипа D в Иордании [15]. T116N, P120S/E, I/T126A/N/I/S, Q129H/R, M133L, K141E, P142S, D144A/E и G145R/A были описаны в качестве значимых мутаций в отношении реактивациии гепатита В при иммуносупрессивной терапии [16]. Замены P120T, Q129H, M133I/T, F/Y134N/L также ассоциированы с ложноотрицательными ошибками диагностических тестов, с ускользанием от поствакцинального ответа или от иммуноглобулиновой терапии [17]. P120T, T126S, Q129H, G130N, S143L, D144A и G145A/R были отмечены как связанные с высоким риском диагностических ошибок [18]. Мутации гена S E164D, I195M, P217L и P120S были выявлены у населения со скрытой инфекцией ВГВ в Северной Бразилии [19]. Особый интерес представляет ген X – в многочисленных работах было показано, что он участвует в канцерогенезе. Ключевые мутации в гене Х, такие как G1613A, C1653T, T1674C/G, T1753V и A1762T/G1764A, постепенно накапливаются в процессе канцерогенеза и могут являться факторами риска развития ГЦК. Присутствие комбинации мутаций гена Х может быть использовано для прогнозирования возникновения и прогрессирования ГЦК [20]. Z. Belaiba и соавт. провели анализ всего генома ВГВ до и во время длительной терапии у 5 пациентов с хронической формой гепатита В и определили мутации в генах S, C и X, которые могут быть связаны с прогрессированием заболевания до ЦП и/или ГЦК [2].
Таким образом, несмотря на то что многие исследования ВГВ были сосредоточены на конкретных областях генома вируса, таких как гены S или X, существует необходимость в полногеномном секвенировании ВГВ, для чего разработаны различные подходы. Типичными методами являются амплификация фрагментов длиной 1000–1500 п.н. и секвенирование по Сэнгеру [2, 21]. Вероятно, из-за трудозатратности по времени и стоимости секвенирования по Сэнгеру в таких исследованиях обычно исследуют относительно небольшое число образцов. Например, Ю.В. Останкова и соавт. определяли полную последовательность генома ВГВ в 3 образцах [22]. В упомянутом выше исследовании Z. Belaiba и соавт. приняли участие 5 пациентов [2]. Также были опубликованы исследования с относительно большим числом образцов. S. Lin и соавт. получили 183 полные последовательности генома ВГВ из 1263 образцов сыворотки крови, положительных на HBsAg [23].
Секвенирования нового поколения (Next generation sequencing, NGS) позволяет проводить анализ большего числа образцов с наименьшими затратами средств и времени по сравнению секвенированием по Сэнгеру. Имеется относительно небольшое количество работ, в которых полный геном ВГВ был определен с помощью метода NGS. Так, Q. Chen и соавт. определили последовательности ВГВ всего генома у 9 пациентов в четырех разных временны́х точках с помощью NGS (амплификация длинных участков ВГВ + Celero EZ DNA-Seq) [24]. S. Lin и соавт. использовали NGS для исследования того, как вирусные квазивиды всего генома ВГВ эволюционируют и диверсифицируются в ответ на сероконверсию HBeAg и вирусный контроль, у 50 пациентов [25]. Мы разработали панель амплификации NGS для быстрого и надежного секвенирования всего генома ВГВ и протестировали ее на 246 клинических образцах.
Материалы и методы
Клинические образцы
Для подбора оптимальных условий пробоподготовки был использован контрольный образец плазмы крови, предоставленный производственным отделом ФБУН ЦНИИ Эпидемиологии Роспотребнадзора. Далее амплификационная панель ВГВ была апробирована на 246 клинических образцах плазмы крови, полученных от пациентов, наблюдающихся по поводу гепатита В в клиническом центре ФБУН ЦНИИ Эпидемиологии Роспотребнадзора. От всех пациентов было получено добровольное информированное согласие на использование образцов плазмы крови в исследовании. Отбор пациентов по возрасту, полу, этнической принадлежности или клиническим особенностям заболевания не проводился. Настоящая работа одобрена локальным Этическим комитетом ФБУН ЦНИИ Эпидемиологии Роспотребнадзора (протокол № 133 от 2 марта 2023 г.). Выделение ДНК из образцов плазмы крови проводили с использованием набора реагентов «РИБО-преп» («АмплиСенс», Россия). Вирусную нагрузку для 210 образцов определяли с помощью набора реагентов HBV-Monitor-FL («АмплиСенс», Россия). Средняя вирусная нагрузка составила 2,81 × 104 МЕ/мл. Вирусная нагрузка для 36 образцов была неизвестна. Информация о поле и возрасте пациентов, а также вирусной нагрузке для каждого образца содержится в таблице S1, представленной в дополнительных материалах на сайте журнала.
Подбор праймеров
В настоящем исследовании в качестве референсной последовательности для подбора праймеров был использован генотип E ВГВ из базы данных HBVdb [26]. Процесс подбора подходящих праймеров повторял метод, представленный в предыдущей публикации [27]. Во-первых, в геноме был сделан выбор предпочтительной для связывания праймера области-мишени в диапазоне 60–150 п.н. Затем данная область была разделена на все возможные фрагменты в диапазоне от 16 до 30 п.н., которые дополнительно отбирали на основе их температуры плавления (Tm), рассчитанной с помощью Thermo Fisher multiple primer analyser. Были выбраны только олигонуклеотиды с Tm между 62 и 64 °C. Затем к выбранным фрагментам добавляли адаптерные последовательности и удаляли любые олигонуклеотиды, которые образовывали гомодимеры. Для оценки потенциальных взаимодействий праймеров были рассчитаны попарные значения изменения свободной энергии Гиббса между каждым олигонуклеотидом и праймерами, включенными в пул ранее. Для расчета изменения свободной энергии Гиббса использовали программу PrimerDimer [28]. В результате, в пул добавляли один олигонуклеотид с наилучшей суммарной оценкой парного изменения свободной энергии Гиббса. Данный процесс повторяли многократно, двигаясь от 5’-конца к 3’-концу генома, пока весь вирусный геном не был покрыт праймерами. В процессе тестирования панели некоторые праймеры были заменены на подобранные вручную. В результате были выбраны 54 праймера для амплификации вирусного генома, разбитого на 20 регионов.
Секвенирование ВГВ
Для амплификации всего генома ВГВ использовали панель праймеров, содержащих на концах последовательности адаптеров Illumina. Мультиплексную полимеразную цепную реакцию (ПЦР) проводили в двух отдельных реакциях, содержащих 10 мкл матричной ДНК, 10 мкл PCR-mix-2-blue («АмплиСенс», Россия), 1,4 мкл dNTP 4,4 мМ («АмплиСенс», Россия), праймеры (конечная концентрация каждого праймера в реакционной смеси и номер пула указаны в таблице S2 в дополнительных материалах на сайте журнала) и стерильную воду, не содержащую нуклеаз, в конечном объеме 25 мкл. В результате при средней вирусной нагрузке 1,12 × 105 MЕ/мл в ПЦР-смеси присутствовало 2,24 × 103 копий. Протокол амплификации: (1) денатурация при 95 °C в течение 3 мин; (2) 16 циклов: 95 °C – 30 с, 55 °C – 30 с, 72 °C – 20 с; (3) финальная элонгация при 72 °C в течение 3 мин. Смешанные продукты ПЦР очищали с использованием магнитных частиц AMPure XP (Beckman Coulter, США) в соотношении 1 : 1, элюция в 15 мкл 0,1× TE. Индексацию ПЦР проводили в реакционном объеме 25 мкл, содержащем 10 мкл PCR-mix-2-blue («АмплиСенс», Россия), 1,4 мкл dNTP 4,4 мм («АмплиСенс», Россия), 5 мкл очищенных продуктов ПЦР, стерильную воду, не содержащую нуклеаз, и адаптеры Nextera index, конечная концентрация каждого праймера составляла 200 нМ. Профиль амплификации: (1) денатурация при 95 °C в течение 1 мин; (2) 25 циклов: 95 °C – 20 с, 55 °C – 30 с, 72 °C – 20 с; (3) финальная элонгация при 72 °C в течение 3 мин. Продукты ПЦР визуализировали с помощью электрофореза в 1,7% агарозном геле, окрашенном бромистым этидием. Смешанные продукты ПЦР-индексации очищали с использованием магнитных частиц AMPure XP (Beckman Coulter, США) в соотношении 1 : 1. Концентрацию очищенной библиотеки измеряли с помощью набора для анализа Qubit dsDNA HS Assay Kit на флуориметре Qubit 4.0 (Invitrogen, США). Высокопроизводительное секвенирование проводили на платформе Illumina MiSeq с использованием набора реагентов MiSeq v2 (300 и 500 циклов) или набора реагентов MiSeq v3 (600 циклов).
Биоинформатический анализ
Полученные прочтения прошли отбор по контролю качества секвенирования с помощью программы FastQC. Последовательности адаптеров были удалены из прочтений с помощью программы Trimmomatic, а последовательности праймеров – с помощью программы Cutadapt. Далее обработанные прочтения были выровнены на референсную последовательность с помощью программного обеспечения Bowtie2. На последнем этапе последовательности были импортированы из файлов формата bam с помощью программы iVar.
Программа для сборки генома ВГВ
Вместе с панелью праймеров мы представляем программу для обработки данных, полученных с помощью нашего метода. Программа основана на нескольких ключевых идеях: 1) рассматриваются только прочтения, содержащие известные праймеры на концах; 2) также прочтения должны быть сходными с известными генотипами ВГВ; 3) качество прямых прочтений обычно лучше, чем качество обратных прочтений, поэтому при перекрывании парных прочтений учитываются прямые прочтения; 4) качество прочтений обычно снижается ближе к концу, поэтому при перекрывании ампликонов предпочтение отдается ампликону с бо́льшим номером. Несмотря на большое количество возражений, представленная схема позволяет быстро и легко генерировать геномы ВГВ из прочтений NGS. Более того, во многих случаях программа позволяет получить правильный результат, в то время как стандартная биоинформатическая обработка приводит к ошибкам, в основном связанным с несоответствием истинной и референсной последовательностей. Используя необработанные данные NGS, программа собирает геномы ВГВ и проводит анализ покрытия и гетерогенности прочтений. Запуск возможен из любой среды разработки Python или из командной строки в любой операционной системе. Программа и подробное описание доступны по адресу https://github.com/ChanyshevMD/HBV_seq.
Валидация панели
Для проверки панели ВГВ мы секвенировали три образца разных генотипов (образец 1: A2; образец 2: D3; образец 3: B2), используя альтернативный подход. ДНК ВГВ амплифицировали с использованием трех пар праймеров AF:
5’-AAGAACTCCCTCGCCTC-3’, AR: 5’-GATGATGGGATGGGAATACARGTG-3’, BF: 5’-GGTATGTTGCCCGTTTGTCC-3’, BR: 5’-GCWAGGAGTTCCGCAGTATGG-3’, CF: 5’-TGCCAAGTGTTTGCTGACGC- 3’, CR: 5’- TGAGATCTTCTGCGACGCGG-3’.
Длины ампликонов для каждой пары праймеров AF/AR, BF/BR и CF/CR составляли соответственно 1460, 829 и 1257 п.н. (изолят P2-121214, GenBank: AB981583.1). Пары праймеров AF/AR и BF/BR были использованы в исследовании Hebeler-Barbosa и соавт. [29]. ПЦР-смесь содержала 10 мкл матричной ДНК, 10 мкл PCR-mix-2-blue («АмплиСенс», Россия), 1,4 мкл dNTP 4,4 мм («АмплиСенс», Россия), праймеры с конечной концентрацией 200 нМ и стерильную воду, не содержащую нуклеаз, в конечном объеме 25 мкл. Протокол амплификации: (1) денатурация при 95 °C в течение 3 мин; (2) 16 циклов: 95 °C – 30 с, 55 °C – 30 с, 72 °C – 90 с; (3) финальная элонгация при 72 °C в течение 3 мин. Длину полученных продуктов ПЦР проверяли с помощью электрофореза в 1% агарозном геле. Очищенные продукты ПЦР количественно определяли с помощью флуориметра Qubit 2.0 (Invitrogen) и набора DNA High Sensitivity Assay kit (ThermoFisher). Для каждого образца три продукта ПЦР (A, B и C) разводили до 0,2 нг/мкл и смешивали в эквимолярной концентрации. Подготовку библиотеки проводили с использованием набора Nextera XT DNA (Illumina) в соответствии с инструкциями производителя, изменив концентрацию вводимой ДНК до 0,5 нг (ампликоны A, B и C для каждого образца). Проиндексированная библиотека Nextera была далее обработана как стандартная библиотека панели ВГВ после индексации. Контроль качества прочтений осуществляли с помощью FastQC. Удаление последовательностей адаптеров из прочтений было произведено с помощью программы Trimmomatic, а удаление последовательностей праймеров – с помощью программы Cutadapt. Далее обработанные прочтения были выровнены на референсную последовательность с помощью программного обеспечения bwa-mem. На последнем этапе последовательности были импортированы из файлов формата bam с помощью программы iVar. Кроме того, нуклеотидные последовательности продуктов ПЦР были определены методом секвенирования по Сэнгеру, которое проводилось в специализированном отделе ФБУН ЦНИИ Эпидемиологии Роспотребнадзора с помощью 3500xL Genetic Analyzer (Applied Biosystems, США) и набора BigDye Terminator v1.1 Cycle Sequencing Kit (ThermoFisher, США). Результаты секвенирования с помощью нашей панели ВГВ, Nextera и по Сэнгеру совпадали практически полностью. Файлы сырых прочтений NGS и по Сэнгеру для всех трех образцов могут быть загружены по ссылке https://github.com/ChanyshevMD/HBV_seq.
Построение филогенетического дерева
Для построения филогенетического дерева были использованы полученные в настоящей работе нуклеотидные последовательности и 199 референсных геномов ВГВ (список S1, см. дополнительные материалы на сайте журнала) из базы данных HBVdb [26], представляющих генотипы A, B, D и E. Множественное выравнивание последовательностей произведено с использованием MAFFT v7.490 [30]. Дерево было построено в MEGA v11.0.11 [31] с использованием алгоритма присоединения соседей и укоренено при помощи метода средней точки. Визуализация выполнена с использованием библиотеки ete3 [32]. Четыре образца (Sample_019, Sample_082, Sample_089, Sample_133) по причине низкого покрытия не были генотипированы и, следовательно, не использовались при нахождении расположения на дереве.
Результаты
Секвенирование клинических образцов
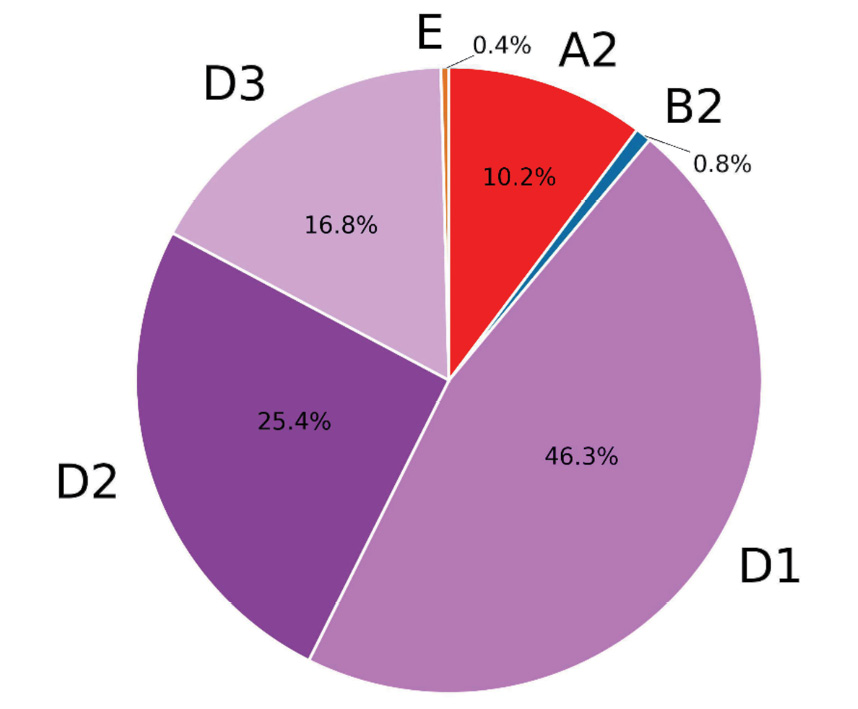
В рамках настоящей работы было секвенировано и проанализировано 246 клинических образцов от пациентов, наблюдающихся по поводу гепатита B в медицинских учреждениях г. Москвы. Глубина прочтения ампликонов представлена на рис. 1. Консенсусные последовательности были собраны при помощи программы, описанной выше, и валидированы путем сравнения с консенсусными последовательностями, полученными стандартными биоинформатическими методами. Полученные последовательности представлены в файле HBV_samples.fa в дополнительных материалах. Средний процент покрытия генома ВГВ составил 96,3%. При этом 126 образцов были прочитаны на 100% (рис. 2). По-видимому, низкий процент покрытия некоторых образцов может быть обусловлен недостаточным количеством и качеством ДНК ВГВ. Генотипы определяли по сходству с референсными геномами, в неоднозначных случаях строили филогенетическое дерево (рис. 3). Большинство образцов (88%) было классифицировано как генотип D. Подтип D1 был установлен в 113 случаях, D2 представлен 62 образцами, а субтип D3 выявлен в 41 исследуемом образце. Всего 11% образцов представляли генотип А, исключительно подтип A2, два исследуемых образца были определены как генотип В2, а в одном случае определен генотип Е. Филогенетическое дерево представлено на рис. 4; следует отметить заметное разнообразие генетических вариантов.

Рис. 1. Log10 глубины прочтения для каждого ампликона, нормированной на общее количество прочтений на образец.
Весь геном ВГВ был поделен на 20 ампликонов (HBV-1–HBV-20).
Fig. 1. Log10 transformed read counts per amplicon norma by total reads per sample.
HBV genome was divided into 20 amplicons (HBV-1–HBV-20).

Рис. 2. Покрытие 246 образцов.
Область генома считали прочитанной при глубине не менее 10 прочтений. Образцы отсортированы по проценту покрытия. Генотипы выделены цветами в соответствии со статьей S. Velkov и соавт. [6].
Fig. 2. Coverage of 246 samples.
The genome region was considered as covered at a minimum depth of 10 reads. Samples are sorted by coverage. The color designation of HBV genotypes corresponds to that from S. Velkov et al. [6].

Рис. 3. Встречаемость различных генотипов ВГВ в Московской области России.
Генотипы выделены цветами в соответствии со статьей S. Velkov и др. [6].
Fig. 3. Occurrence of various HBV genotypes in the Moscow region of Russia.
The color designation of HBV genotypes corresponds to that from [6].

Рис. 4. Филогенетическое дерево секвенированных образцов ВГВ.
Дерево было построено в MEGA v11.0.11 с использованием алгоритма присоединения соседей и укоренено при помощи метода средней точки. Визуализация выполнена с использованием библиотеки ete3. Генотипы A, B, D, E отмечены соответственно красным, синим, фиолетовым и оранжевым цветами. Референсные последовательности помечены черным цветом, а секвенированные в данной работе – желтым.
Fig. 4. Phylogenetic tree for sequenced HBV samples.
The tree was constructed in MEGA v11.0.11 using the neighbor joining algorithm and rooted using the midpoint method. Visualization was made using the ete3 library. Genotypes A, B, D, E are marked in red, blue, violet, and orange, respectively. References and sequences from this study are highlighted in black and yellow, respectively.
Последующий анализ секвенированных последовательностей включал выявление и оценку распространенности мутаций ВГВ, значимых в отношении терапии и профилактики заболевания. К подобного рода мутациям относятся замены в RT-домене гена P: L80I/V, I169T, V173L/M, L180M, A181S, T184I/L/S, S202G, M204V/I, N236T, M250V, ассоциированные с устойчивостью к противовирусной терапии, а также такие замены в гене S, как L109R, T116N, P120S/T/E, T126A/N/I/S, Q129H/R, G130N, M133L/I/T, F/Y134N/L, K141E/I/R, P142S, S/T143L, D144A/E, G145A/R, E164D, I195M, связанные с иммунным ускользанием. Пять образцов содержали по меньшей мере одну мутацию, связанную с устойчивостью к противовирусной терапии, и в 23 образцах была найдена по меньшей мере одна мутация, связанная с ускользанием от иммунного ответа. Кроме того, в 8 образцах находилась по меньшей мере одна аминокислотная замена в местах мутаций, связанных с ускользанием от иммунитета. Мутации представлены в виде диаграммы на рис. 5. Не менее важным вопросом в научной деятельности является поиск и углубленное изучение мутаций вируса, оказывающих значимое влияние на прогноз основного заболевания и скорость развития патологии печени. По результатам многочисленных исследований, направленных на формирование представления о специфичных маркерах, характеризующих потенциально неблагоприятное течение гепатита В, основное внимание обращено на X, precore- и core-гены ВГВ. Так, замены W28* и G29D в precore-регионе были выявлены в 69 и 33% образцов соответственно. Аминокислотные замены F24Y, E64D, E77Q, A80I/T/V, L116I и cE180A в core-регионе встречались в 10,5, 22, 10, 38, 44 и 0,4% образцов соответственно. В гене X ВГВ нуклеотидные замены G1613A, C1653T, T1674S, T1753V, A1762T, G1764A и T1768A обнаружены в 9, 11, 11, 26, 26, 33 и 2 образцов соответственно. Данные замены, согласно данным литературы [20, 33], являются маркерами, указывающими на вероятность неблагоприятного течения гепатита В и высокую скорость развития ГЦК. Распространенность вышеуказанных мутаций ВГВ сравнительно велика в исследуемой выборке пациентов Московского региона. Данный факт, несомненно, указывает на необходимость дальнейшего мониторинга вируса, циркулирующего на территории региона, с целью формирования прогностических моделей и оптимизации оказания медицинской квалифицированной помощи.

Рис. 5. Встречаемость мутаций в RT-домене гена P, связанных с устойчивостью к противовирусной терапии, и мутаций в гене S, связанных с ускользанием от поствакцинального ответа.
Серые поля представляют собой неопределенные области, белые – дикий тип, желтые – замены, не описанные в литературе, красные – мутации, описанные в литературе как мутации устойчивости к противовирусной терапии и мутации, ускользания от иммунитета. Представлено всего 33 образца, содержащих хотя бы одно отличие от дикого типа.
Fig. 5. The detection rates of mutations in RT domain of P gene associated with resistance to antiviral therapy and mutations in S gene associated with evasion of vaccine-induced response.
Gray, white, yellow, and red boxes represent respectively the undetermined regions, wild type, substitutions not described in the literature, and substitutions described in the literature data as associated with resistance to antiviral therapy and vaccine escape mutations. Total 33 samples containing at least one difference from the wild type are presented.
Обсуждение
Полученные в настоящем исследовании данные о генотипическом разнообразии ВГВ, представленном на территории Московского региона, согласуются с немногочисленными опубликованными результатами коллег. Так, в работе В.В. Клушкиной и соавт., по результатам проведенного секвенирования области гена S в 63 образцах сыворотки крови из Московской области, генотип D был обнаружен в 84% исследованных образцов (52/63), генотип А – в 14% (9/63) и генотип С – в 2% (1/63). Кроме того, генотип D был представлен четырьмя подтипами: D1, D2, D3 и D4; генотип A состоял только из подтипа A2, а генотип C – только из подтипа C1 [34]. Согласно исследованию В.А. Мануйлова и соавт., распределение генотипов ВГВ в выборках городского населения бывшего западного СССР (179 человек) представляет собой: D – 82%, A – 17%, C – 1%. Генотип D был представлен подтипами D1, D2 и D3, генотип A – подтипом А2, генотип C – подтипом C2 [35]. Кроме того, наши данные согласуются с обзорной статьей о распространенности генотипов ВГВ во всем мире [6]. Сравнение распространенности мутаций, связанных с резистентностью к противовирусной терапии или ускользанием от поствакцинального ответа, затруднено из-за различий в терапевтических подходах в разных странах. Например, в России почти все население вакцинировано против ВГВ, но при этом противовирусная терапия АН практически не применяется. В упомянутой выше статье В.А. Мануйлова и соавт. описаны нуклеотидные последовательности ВГВ городского населения бывшего западного СССР, 2 из 179 образцов содержат по меньшей мере одну мутацию, связанную с резистентностью к терапии (rtL173V KX925288 и rtS202G AY603465), в 14 образцах обнаружена по меньшей мере одна мутация, связанная с устойчивостью к иммунитету. Кроме того, 8 образцов содержали по меньшей мере одну аминокислотную замену в местах мутаций, возникающих в вирусных геномах в ответ на вакцинацию. Мутации представлены в виде диаграммы на рисунке S1 (см. дополнительные материалы). Таким образом, встречаемость мутаций в образцах из работы В.А. Мануйлова и соавт. согласуется с результатами, полученными в настоящем исследовании: несколько мутаций устойчивости к противовирусной терапии и около 20 мутаций, связанных с ускользанием от иммунного ответа, среди двух сотен образцов.
Мутации X, precore- и core-регионов ВГВ, охарактеризованные в научной мировой литературе в качестве маркеров неблагоприятного течения и исходов заболевания, также затронуты в настоящей работе в силу важности изучения функциональных замен в данных регионах и относительно высокой распространенности этих замен. Проведение подобного рода исследований, направленных на пристальное изучение отдельных мутаций ВГВ и оказываемого эффекта на организм человека, безусловно, требует быстрого и в то же время точного метода определения нуклеотидной последовательности циркулирующего возбудителя. Полученные в настоящей работе результаты указывают на возможность применения амплификационной панели с целью проведения полногеномного секвенирования ВГВ при помощи NGS и последующего анализа. Сферы применения получаемых данных охватывают области научных интересов как со стороны классической эпидемиологии, так и с точки зрения развития персонифицированного подхода к ведению пациента. С одной стороны, применение методов полногеномного секвенирования позволит использовать получаемые данные в рутинной научной практике при проведении эпидемиологических исследований по оценке и анализу распространенности генотипов и субтипов вируса на территории РФ, при формировании прогностических эпидемиологических моделей развития эпидемического процесса, проведении эпидемиологических расследований и др. С другой стороны, данный подход позволит решить узкие научные задачи по выявлению нуклеотидных замен – как активно изучаемых в мире, так и впервые обнаруживаемых.
Заключение
В настоящей работе детально изложены этапы проведения полногеномного секвенирования ВГВ, приведены лабораторный протокол, нуклеотидные последовательности используемых праймеров и подход к анализу полученных данных. На примере выборки клинических образцов показана состоятельность применяемой панели в частности и применяемого метода в целом. Методология, продемонстрированная в настоящем исследовании, обладает большим потенциалом для использования для научных и практических целей. Данный подход может широко применяться в исследовательских проектах для углубления знаний о ВГВ и выявления сложных взаимосвязей между генетическими вариантами возбудителя и клиническими исходами заболевания. Кроме того, полногеномное секвенирование ВГВ может быть использовано в рамках оптимизации системы эпидемиологического надзора, позволяя обеспечивать эффективный мониторинг за динамикой распространения возбудителя. Примечательно, что понимание динамики мутационного процесса вирусного генома и закрепления в популяции определенных вариантов в конечном итоге может обеспечить реализацию программ по персонализированной терапии, позволяя принимать обоснованные решения с учетом конкретных генетических профилей ВГВ у пациентов.
Об авторах
Михаил Дамирович Чанышев
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Автор, ответственный за переписку.
Email: chanishq@gmail.com
ORCID iD: 0000-0002-6943-2915
канд. биол. наук, научный сотрудник Лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. МоскваНаталья Викторовна Власенко
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: vlasenko@cmd.su
ORCID iD: 0000-0002-2388-1483
научный сотрудник Лаборатории вирусных гепатитов ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. МоскваГерман Викторович Роев
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора; Московский физико-технический институт
Email: roevherman@gmail.com
ORCID iD: 0000-0002-2353-5222
bioinformatician of the Laboratory for genomic research, Central Research Institute for Epidemiology of the Federal Service for Surveillance of Consumer Rights Protection and Human Wellbeing
Россия, 111123, г. Москва; 141701, г. ДолгопрудныйИван Андреевич Котов
Московский физико-технический институт
Email: ivan.kotov@phystech.edu
ORCID iD: 0000-0003-2416-5689
аспирант ФБМФ МФТИ
Россия, 141701, г. ДолгопрудныйАльбина Григорьевна Глущенко
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора; Московский физико-технический институт
Email: albinagluschenko@gmail.com
ORCID iD: 0009-0002-8851-8703
лаборант Лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. Москва; 141701, г. ДолгопрудныйВера Васильевна Макашова
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: veramakashova@yandex.ru
ORCID iD: 0000-0002-0982-3527
доктор медицинских наук, профессор, ведущий научный сотрудник клинического отдела инфекционной патологии ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. МоскваКамиль Фаридович Хафизов
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: kkhafizov@gmail.com
ORCID iD: 0000-0001-5524-0296
канд. биол. наук, заведующий Лабораторией геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. МоскваВасилий Геннадьевич Акимкин
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
академик РАН, доктор медицинских наук, профессор, директор ФБУН ЦНИИ Эпидемиологии Роспотребнадзора
Россия, 111123, г. МоскваСписок литературы
- WHO. Hepatitis B; 2022. Available at: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b
- Belaiba Z., Ayouni K., Gdoura M., Kammoun Rebai W., Touzi H., Sadraoui A., et al. Whole genome analysis of hepatitis B virus before and during long-term therapy in chronic infected patients: Molecular characterization, impact on treatment and liver disease progression. Front. Microbiol. 2022; 13: 1020147. https://doi.org/10.3389/fmicb.2022.1020147
- Kramvis A. Genotypes and genetic variability of hepatitis B virus. Intervirology. 2014; 57(3-4): 141–50. https://doi.org/10.1159/000360947
- Tran T.T., Trinh T.N., Abe K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008; 82(11): 5657–63. https://doi.org/10.1128/JVI.02556-07
- Tatematsu K., Tanaka Y., Kurbanov F., Sugauchi F., Mano S., Maeshiro T., et al. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009; 83(20): 10538–47. https://doi.org/10.1128/JVI.00462-09
- Velkov S., Ott J.J., Protzer U., Michler T. The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes (Basel). 2018; 9(10): 495. https://doi.org/10.3390/genes9100495
- Sunbul M. Hepatitis B virus genotypes: global distribution and clinical importance. World J. Gastroenterol. 2014; 20(18): 5427–34. https://doi.org/10.3748/wjg.v20.i18.5427
- Delaney W.E. 4th., Yang H., Westland C.E., Das K., Arnold E., Gibbs C.S., et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003; 77(21): 11833–41. https://doi.org/10.1128/jvi.77.21.11833-11841.2003
- Zhang X., Chen X., Wei M., Zhang C., Xu T., Liu L., et al. Potential resistant mutations within HBV reverse transcriptase sequences in nucleos(t)ide analogues-experienced patients with hepatitis B virus infection. Sci. Rep. 2019; 9(1): 8078. https://doi.org/10.1038/s41598-019-44604-6
- Vincenti D., Piselli P., Solmone M., D’Offizi G., Capobianchi M.R., Menzo S. Evolutionary trends of resistance mutational patterns of HBV reverse transcriptase over years (2002–2012) of different treatment regimens: The legacy of lamivudine/adefovir combination treatment. Antiviral. Res. 2017; 143: 62–8. https://doi.org/10.1016/j.antiviral.2017.03.008
- Araujo N.M., Teles S.A., Spitz N. Comprehensive analysis of clinically significant hepatitis B virus mutations in relation to genotype, subgenotype and geographic region. Front. Microbiol. 2020; 11: 616023. https://doi.org/10.3389/fmicb.2020.616023
- Liu Y., Wang C., Zhong Y., Li X., Dai J., Ren X., et al. Genotypic resistance profile of hepatitis B virus (HBV) in a large cohort of nucleos(t)ide analogue-experienced Chinese patients with chronic HBV infection. J. Viral. Hepat. 2011; 18(4): e29–39. https://doi.org/10.1111/j.1365-2893.2010.01360.x
- Carman W.F. The clinical significance of surface antigen variants of hepatitis B virus. J. Viral. Hepat. 1997; 4(Suppl. 1): 11–20. https://doi.org/10.1111/j.1365-2893.1997.tb00155.x
- Mokaya J., Vasylyeva T.I., Barnes E., Ansari M.A., Pybus O.G., Matthews P.C. Global prevalence and phylogeny of hepatitis B virus (HBV) drug and vaccine resistance mutations. J. Viral. Hepat. 2021; 28(8): 1110–20. https://doi.org/10.1111/jvh.13525
- Ababneh N.A., Sallam M., Kaddomi D., Attili A.M., Bsisu I., Khamees N., et al. Patterns of hepatitis B virus S gene escape mutants and reverse transcriptase mutations among genotype D isolates in Jordan. PeerJ. 2019; 7: e6583. https://doi.org/10.7717/peerj.6583
- Lazarevic I., Banko A., Miljanovic D., Cupic M. Immune-escape hepatitis B virus mutations associated with viral reactivation upon immunosuppression. Viruses. 2019; 11(9): 778. https://doi.org/10.3390/v11090778
- Avellón A., Echevarria J.M. Frequency of hepatitis B virus ‘a’ determinant variants in unselected Spanish chronic carriers. J. Med. Virol. 2006; 78(1): 24–36. https://doi.org/10.1002/jmv.20516
- Ma Q., Wang Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J. Med. Virol. 2012; 84(2): 198–206. https://doi.org/10.1002/jmv.23183
- Araújo S.D.R., Malheiros A.P., Sarmento V.P., Nunes H.M., Freitas P.E.B. Molecular investigation of occult hepatitis B virus infection in a reference center in Northern Brazil. Braz. J. Infect. Dis. 2022; 26(3): 102367. https://doi.org/10.1016/j.bjid.2022.102367
- Zhou X., Liu D., Li Z., Zhao J., Cai S., Cao G. The mechanism of hepatitis B virus X gene in promoting hepatocellular carcinoma. J. Cancer Sci. Clin. Ther. 2022; 6(2): 222–33. https://doi.org/10.26502/jcsct.5079158
- Abdou Chekaraou M., Brichler S., Mansour W., Le Gal F., Garba A., Dény P., et al. A novel hepatitis B virus (HBV) subgenotype D (D8) strain, resulting from recombination between genotypes D and E, is circulating in Niger along with HBV/E strains. J. Gen. Virol. 2010; 91(Pt. 6): 1609–20. https://doi.org/10.1099/vir.0.018127-0
- Останкова Ю.В., Семенов А.В., Зуева Е.Б., Тотолян А.А. Первые случаи выявления вируса гепатита B субгенотипа D4 у больных хроническим, острым и скрытым вирусным гепатитом B в Российской Федерации. Молекулярная генетика, микробиология и вирусология. 2020; 38(4): 180–7. https://doi.org/10.17116/molgen202038041180 https://elibrary.ru/cuboiu
- Liu H., Shen L., Zhang S., Wang F., Zhang G., Yin Z., et al. Complete genome analysis of hepatitis B virus in Qinghai-Tibet plateau: the geographical distribution, genetic diversity, and co-existence of HBsAg and anti-HBs antibodies. Virol. J. 2020; 17(1): 75. https://doi.org/10.1186/s12985-020-01350-w
- Chen Q.Y., Jia H.H., Wang X.Y., Shi Y.L., Zhang L.J., Hu L.P., et al. Analysis of entire hepatitis B virus genomes reveals reversion of mutations to wild type in natural infection, a 15 year follow-up study. Infect. Genet. Evol. 2022; 97: 105184. https://doi.org/10.1016/j.meegid.2021.105184
- Lin S.R., Yang T.Y., Peng C.Y., Lin Y.Y., Dai C.Y., Wang H.Y., et al. Whole genome deep sequencing analysis of viral quasispecies diversity and evolution in HBeAg seroconverters. JHEP Rep. 2021; 3(3): 100254. https://doi.org/10.1016/j.jhepr.2021.100254
- HBVdb. Available at: https://hbvdb.lyon.inserm.fr/HBVdb/HBVdbIndex
- Kotov I., Saenko V., Borisova N., Kolesnikov A., Kondrasheva L., Tivanova E., et al. Effective approaches to study the genetic variability of SARS-CoV-2. Viruses. 2022; 14(9): 1855. https://doi.org/10.3390/v14091855
- Johnston A.D., Lu J., Ru K.L., Korbie D., Trau M. PrimerROC: accurate condition-independent dimer prediction using ROC analysis. Sci. Rep. 2019; 9(1): 209. https://doi.org/10.1038/s41598-018-36612-9
- Hebeler-Barbosa F., Wolf I.R., Valente G.T., Mello F.C.D.A., Lampe E., Pardini M.I.M.C., et al. A new method for next-generation sequencing of the full hepatitis B virus genome from a clinical specimen: impact for virus genotyping. Microorganisms. 2020; 8(9): 1391. https://doi.org/10.3390/microorganisms8091391
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30(4): 772–80. https://doi.org/10.1093/molbev/mst010
- Tamura K., Stecher G., Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021; 38(7): 3022–7. https://doi.org/10.1093/molbev/msab120
- Huerta-Cepas J., Serra F., Bork P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016; 33(6): 1635–8. https://doi.org/10.1093/molbev/msw046
- Al-Qahtani A.A., Al-Anazi M.R., Nazir N., Abdo A.A., Sanai F.M., Al-Hamoudi W.K., et al. The correlation between hepatitis B virus precore/core mutations and the progression of severe liver disease. Front. Cell Infect. Microbiol. 2018; 8: 355. https://doi.org/10.3389/fcimb.2018.00355
- Klushkina V.V., Kyuregyan K.K., Kozhanova T.V., Popova O.E., Dubrovina P.G., Isaeva O.V., et al. Impact of universal hepatitis В vaccination on prevalence, infection-associated morbidity and mortality, and circulation of immune escape variants in Russia. PLoS One. 2016; 11(6): e0157161. https://doi.org/10.1371/journal.pone.0157161
- Manuylov V., Chulanov V., Bezuglova L., Chub E., Karlsen A., Kyuregyan K., et al. Genetic diversity and possible origins of the hepatitis B virus in Siberian natives. Viruses. 2022; 14(11): 2465. https://doi.org/10.3390/v14112465
Дополнительные файлы

