



Bastroviruses (Astroviridae): genetic diversity and potential impact on human and animal health
- Authors: Roev G.V.1,2, Borisova N.I.1, Chistyakova N.V.3, Vyhodtseva A.V.1, Akimkin V.G.1, Khafizov K.F.1
-
Affiliations:
- Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing
- Moscow Institute of Physics and Technology (National Research University)
- A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences
- Issue: Vol 68, No 6 (2023)
- Pages: 505-512
- Section: ORIGINAL RESEARCHES
- URL: https://virusjour.crie.ru/jour/article/view/14736
- DOI: https://doi.org/10.36233/0507-4088-192
- EDN: https://elibrary.ru/iqblwy
- ID: 14736

Cite item

Abstract
Introduction. Bastroviruses were discovered in the Netherlands in 2016 in human stool samples and show partial genetic similarities to astroviruses and hepatitis E viruses. Their association with disease onset has not yet been established.
Materials and methods. Metagenomic sequencing of fecal samples of Nyctalus noctula bats collected in the Russian Federation in 2023 was performed. Two almost complete genomes of bastroviruses were assembled. The zoonotic potential of these viruses was assessed using machine learning methods, their recombination was studied, and phylogenetic trees were constructed.
Results. A nearly complete bastrovirus genome was de novo assembled in one of the samples, and it was used to assemble another genome in another sample. The zoonotic potential of the virus from one of these samples was estimated as high. The existence of recombination between structural and non-structural polyproteins was demonstrated.
Conclusion. Two bastrovirus genomes were assembled, phylogenetic and recombination analyses were performed, and the zoonotic potential was evaluated.
Keywords
Full Text
Introduction
In 2016, a previously unknown RNA virus was found during metagenomic sequencing of 200 human fecal samples in the Netherlands [1]. A new pathogen with a certain genetic similarity to astrovirus and hepatitis E was identified in 7 of these samples. Sequence lengths ranged from 6017 to 6339 nucleotide bases. The virus had the same genome structure in all 7 samples and the amino acid sequence similarity of the putative nonstructural ORF1 protein ranged from 67 to 93%. Likewise, the similarity of the putative structural protein ORF2 ranged from 73 to 98%. This high degree of diversity allowed the authors of this article to hypothesize that bastrovirus as a human infectious agent has likely been circulating among humanity for a long time instead of having emerged recently. Such diversity may also be related to emergence in different reservoirs and transmission to humans through contaminated food or in the form of a zoonotic infection from domestic animals, livestock or wildlife. All bastrovirus genomes contained similar conserved amino acid domains. The capsid protein of bastroviruses showed remarkable variability, especially in the initial 40-nucleotide N-terminal and final 242-nucleotide C-terminal sequences. Several antigenic epitopes with a length of more than ten amino acids were detected at the C-terminus of the capsid protein. A nested polymerase chain reaction (PCR) assay targeting the 5’-region of the genome identified the bastrovirus in 32 of the 200 fecal samples tested. However, there was no clear correlation found between clinical symptoms such as diarrhea and the presence of the virus [1].
Since the initial detection of bastrovirus in human feces in 2016, it has been repeatedly identified in other various samples. In 2017, a bastrovirus with a genome size of 5875 bases was identified in Brazilian wastewater. Its genome similarity was 56% in comparison to previously known bastroviruses (GenBank: KX907135) [2]. The researchers hypothesized that its reservoir could be mammalian. In a 2018 study, a bastrovirus (GenBank: MG693175) was identified in 87 fecal samples from bats of two different species in Cameroon [3]. In a 2019 study during a swine diarrhea outbreak in the United States [4], a bastrovirus genome of 5881 bases in length with 84% similarity to swine bastrovirus (GenBank: KX907134) was detected in fecal samples and subsequently named PBastV-USA 2017-1. Sequence analysis of its genome revealed 97 and 87% sequence similarity to the ORF1 and ORF2 sequences of the detected bastrovirus, respectively, indicating significant genetic similarity to porcine bastrovirus. These full genome sequence data enabled the development of a quantitative PCR-based test for pathogen detection. In June 2017, this test was used to examine 368 swine samples submitted to the South Dakota State University Animal Disease Laboratory. The bulk of these (90%) were oral swabs and the remainder (10%) were fecal, rectal, or environmental samples. Out of the 368 samples tested, 114 (or 31%) were positive for the virus. However, most of the samples were from healthy pigs because they were collected as part of a farm animal disease surveillance initiative. Thus, the detected bastrovirus was not associated with any specific disease.
An article published in 2019 examined 72 bat specimens of four species caught in 2012 in the city of Bisha and its vicinity in Saudi Arabia [5]. Two species of bats were found to have a virus similar to bastrovirus, but the viruses found in Saudi Arabia were closer to hepeviruses than to the human bastrovirus, with a 50% similarity in amino acid sequence to the former. As a result, the researchers classified them as Middle East Hepe-Astrovirus, as they showed about 70% similarity to bat bastrovirus and rat bastrovirus from Vietnam.
In 2021, Japanese researchers identified nearly complete genome sequences of four bastroviruses found in the feces of healthy pigs [6]. It was found that bastroviruses present in pigs and other animals, including humans, have similar genomic organization. Specifically, they all have three conserved domains: viral methyltransferase, RNA helicase and RNA-dependent RNA polymerase (RdRp) in the non-structural ORF1 and the capsid domain of astrovirus in the structural ORF2. Amino acid sequence comparison showed that the resulting bastroviruses had 95–99% and 76–96% similarity in the ORF1 and ORF2 regions, respectively. However, when pig bastroviruses were compared with bastroviruses from other animals, the similarity was only 21–43% and 9–21% for the ORF1 and ORF2 regions, respectively. This indicates that although the bastroviruses have possibly shared a common ancestor, they evolved separately in each host group over a long period of time. The presence of potential recombination events in the genome also indicates that bastroviruses acquire genetic diversity through recombination events.
In 2023, an article [7] was published and it focused on the insufficiently studied viral diversity of lower vertebrates, including fish, amphibians and reptiles. The object of study was the Asian toad inhabiting China. As a result of those studies, more than 20 new RNA viruses were identified in samples from this toad species. Thus, the nearly complete genome of the bastrovirus was revealed, which is very different from the previously known variants, which allowed us to identify new branches on the phylogenetic tree. This genome, designated as AtBastV/GCCDC11/2022, consists of three putative protein-coding regions, each with varying degrees of similarity to various known viruses. Upon deeper examination of the RdRp phylogeny and capsid segments of AtBastV/GCCDC11, it was found to have strong genetic similarity to a strain of amphibian bastrovirus from the genus Rana. In addition, this strain had marked similarity with astrovirus 2 found in the Hainan black-eyed toad and certain animal astroviruses, although it was distantly related to hepeviruses.
Phylogenetic analysis based on RdRP sequence shows that bastroviruses and hepeviruses form a separate clade on the phylogenetic tree, distinct from astroviruses [3]. Most animal bastroviruses form a monophyletic group and are clustered according to the host species they infect. An exception to this is the Bat_Bastrovirus-like_virus/VietNam/Bat/17819_21 lineage, which forms a distinct outgroup on the phylogenetic tree. Phylogenetic analysis based on capsid protein yields similar results. In a 2016 paper [1] it was hypothesized that recombination had occurred in the past between the genome regions encoding capsid protein and polymerase. As evidence, the authors point to the fact that phylogenetically the capsid protein is more related to astroviruses and the polymerase gene to the hepatitis E virus. Additional recombination events are also indicated by the fact that the CMR/Bat/P24 strain clusters with two bat strains on the polymerase phylogenetic tree and only one strain on the capsid protein phylogenetic tree [3].
Bastroviruses can be differentiated on the basis of genetic traits. ORF1 of all mammalian bastroviruses encodes a viral methyltransferase, followed by a viral helicase and RdRp, whereas ORF2 encodes a capsid protein. ORF3 is not present in the genome of all bastroviruses, and if present, as in the case of human bastroviruses, it encodes the papain-like cysteine protease of hepatitis E virus. Although astroviruses and bastroviruses share some similarities in structural protein, the structure of the bastrovirus genome and its features may contribute to the future classification of these viruses into a new viral family. Furthermore, the extensive genetic diversity among bastroviruses derived from different host species may warrant further classification of bastroviruses into different lineages or genera.
The sources discussed above allow us to come to the conclusion that that bastroviruses are very widespread in humans and various animal species. Bastroviruses have been detected in humans, pigs, bats, river mollusks, toads and even in sewage. This diversity of virus carriers supports the theory that this virus has been circulating in nature for a certain period of time rather than having originated recently. So far, no cases of bastroviruses detected in Russia have been described in any literature. In the present study, we performed metagenomic sequencing of the genetic material of fecal samples of bats of the species Nyctalus noctula from the Saratov Region, which resulted in the discovery of a previously unknown bastrovirus and the assembly of its complete genome. We annotated the new bastrovirus genome, constructed phylogenetic trees and performed recombination analysis.
Materials and methods
Metagenomic sequencing of fecal samples of N. noctula bats collected in February 2023 in Russia was performed. RNA isolation from samples was performed using the QIAamp Viral RNA kit (Qiagen, Germany). Reverse transcription was performed using the REVERTA-L kit (AmpliSens, Russia) according to the manufacturer’s instructions. Second strand DNA synthesis was performed using the NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module kit (E6111L). Purified double-stranded cDNA was fragmented in microTUBE-50 AFA Fiber Screw-Cap (PN 520166) on the Covaris M220 instrument (Covaris, Woburn, MA) up to ~ 550 bp. Libraries for paired-end sequencing were constructed using the NEBNext Ultra End Repair/dA-Tailing Module (NEB E7546L), NEBNext Ultra Ligation Module (NEB E7595L), and Y-adapter compatible with IDT for Illumina Nextera DNA UD Indexes kits. Index PCR was performed using NEBNext Ultra II Q5 Master Mix (NEB #M0492). The final library was validated on an Agilent Bioanalyzer 2100 instrument (Agilent Technologies, USA). Sequencing was performed on MiSeq and NextSeq 2000 platforms (Illumina, USA).
Assembling consensus sequences
In “raw” reads, adapters were removed using the option in Trimmomatic v0.39 ILLUMINACLIP [8]. The options LEADING:7, TRAILING:7, SLIDINGWINDOW:4:20, and MINLEN:40 were also applied. Paired reads were merged using BBmerge [9] (maxstrict=t), and files with unpaired reads for each sample were merged into one. Thus, three files were obtained for each sample: two files with paired-end reads and one file with unpaired reads. Reads corresponding to the host were removed by aligning to the reference genomes of the bats Pipistrellus kuhlii (GCF_014108245.1) and Myotis myotis (GCF_014108235.1) using bowtie2 (options -un and -un-conc separately for unpaired and paired-end reads, respectively). The genomes of these two species were used, as the reference genome for N. noctula is not available in the available databases. Then, using Kaiju v1.9.2 [10] (with the use of nr_euk database), we left only reads either defined as viral or unclassifiable (files with paired-end reads and unpaired reads were once again processed separately). The reads remaining after this filtering were assembled de novo into contigs using MEGAHIT v1.2.9 [11], and the resulting contigs were used to search the NCBI nr database using DIAMOND [12] blastx (options -very-sensitive, e-value 1e-08, -k 3).
In the sample N.noctula_3 (MiSeq), a long (5,832 bp) contig related to bastroviruses was found based on a homology search. This contig was subsequently used as a reference for consensus assembly for another sample. Bastrovirus contigs were also detected in bat fecal samples of N.noctula_4 (NextSeq), N.noctula_4 (MiSeq, technical replicate of the previous sample). Kaiju-filtered reads from the respective samples were aligned with the help of bowtie2 v2.4.4 [13] (--local) to the above reference. “Bam” replica files were merged using the “samtools merge” [14] v1.15.1 command, consensus assembly was performed using the “samtools consensus” command (-m simple -aa -c 0.51). As a result, two consensus sequences were obtained (samples #3, 4).
Phylogenetic analysis
Open reading frames were identified in the assembled sequences using NCBI ORFfinder [15] (option “ATG” and alternative initiation codons) and BLASTX [16]. Amino acid sequences corresponding to non-structural polyprotein (NSP) and structural polyprotein (SP) were used for further phylogenetic analysis. For tree construction we used 31 genome sequences of bastroviruses from NCBI, in which NSP and SP polyproteins were annotated. The list of accession numbers of the samples used is as follows: KU318321.1, NC_035758.1, KU318317.1, KU318320.1, KU318315.1, KU318318.1, KU318318.1, KU318319.1, KU318316.1, KX907134.1, NC_032423.1, MK387176. 1, KX907130.1, MT549856.1, LC549662.1, MF042208.1, KX907133.1, NC_035471.1, OM104033.1, OQ835729.1, KX907129.1, KX907131.1, NC_032484. 1, KX907132.1, KX907128.1, KX907127.1, MG693175.1, MT549857.1, KX907135.1, NC_032426.1, OQ835730.1, MT766313.1. The sequence of goose astrovirus (GenBank: OM200916.1) was used as an outgroup. Alignment was performed using the MAFFT v7.490 program [17]. Tree construction was performed in IQ-TREE v2.2.3 [18] (option -alrt 1000), the optimal model was determined using ModelFinder [19]. Visualization of the tree was performed using the iTOL online tool [20].
Recombination analysis
To investigate potential recombination, multiple alignments to the amino acid sequences of NSP and SP proteins for 33 samples (31 obtained from NCBI and 2 in this research) were constructed in the MAFTT program and then reverse-translated into nucleotide alignments using PAL2NAL [21]. Next, the alignments for NSP and SP proteins were combined. A phylogenetic compatibility matrix with the Robinson-Folds metric was constructed using the RDP4 protocol. The window width was 400, the step was 50 nucleotides [22].
Analysis of zoonotic potential
A developed machine learning model [23], available at https://github.com/Nardus/zoonotic_rank, was used to estimate the zoonotic potential of the bastrovirus. Two bastrovirus genomes and an annotation table were fed as input. Using the PredictNovel.R command, files with the results were obtained.
Results
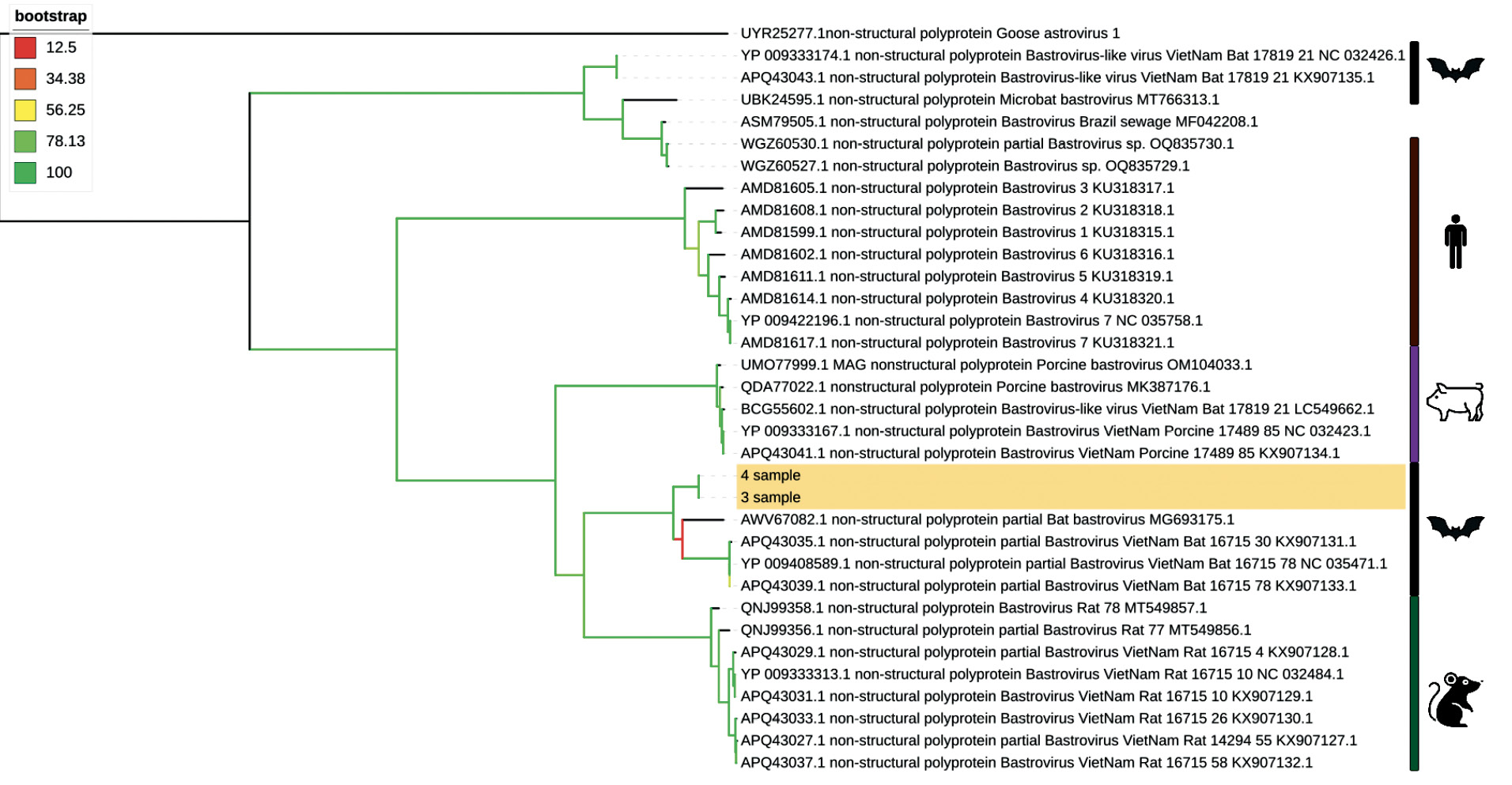
In the present study, nearly complete bastrovirus genome sequences were obtained for two samples based on metagenomic sequencing data from the fecal genetic material of bats N. noctula caught in the territory of the Russian Federation in 2023. A de novo long contig (about 5800 bp) covering the nearly complete bastrovirus genome was assembled in one of the samples, which was then used as a reference to assemble the bastrovirus genome from the other sample. After the annotation of the genomes by searching with the BLASTX programs in the NCBI nr database and the NCBI ORFfinder tool, an amino acid sequence of NSP of about 1200 amino acids in length and SP of about 630 amino acids in length was obtained for the two samples. Phylogenetic analysis performed on the non-structural (Fig. 1) and structural (Fig. 2) polyproteins showed similarity to bastrovirus samples from bats caught in Vietnam. According to the results obtained using the BLASTP program, the similarity to already known sequences is less than 77% for the NSP protein and less than 63% for the SP protein, suggesting that viruses from these samples are novel.

Fig. 1. Phylogenetic tree of the non-structural polyprotein (NSP) of bastroviruses.
Samples sequenced as part of this study are highlighted in yellow. Branch colors indicate node support calculated by the SH-aLRT method.
Рис. 1. Филогенетическое дерево, построенное на основе аминокислотной последовательности неструктурного полипротеина (NSP) бастровирусов.
Образцы, отсеквенированные в рамках этого исследования, выделены желтым. Цветом показана поддержка узлов, рассчитанная по методу SH-aLRT.

Fig. 2. Phylogenetic tree of the structural polyprotein (SP) of bastroviruses.
Samples sequenced as part of this study are highlighted in yellow. Branch colors indicate node support calculated by the SH-aLRT method.
Рис. 2. Филогенетическое дерево, построенное на основе аминокислотной последовательности структурного полипротеина (SP) бастровирусов.
Образцы, отсеквенированные в рамках этого исследования, выделены желтым. Цветом показана поддержка узлов, рассчитанная по методу SH-aLRT.
Study of recombination in bastrovirus genomes
Analysis of bastrovirus genome sequences using the phylogenetic compatibility matrix (Fig. 3) indicates the presence of possible recombination between the reading frames encoding SP and NSP polyproteins, which is generally typical for astroviruses [24, 25], the closest relatives of which are bastroviruses. This is also confirmed by the different topology of phylogenetic trees constructed for SP and NSP proteins. For example, the sample KX907131.1 presented in Fig. 1 is located in the same clade with the samples obtained in the present study, while in Fig. 2, the sample is shown in another clade of bat bastroviruses.

Fig. 3. Phylogenetic compatibility matrix constructed from the merged alignments of NSP and SP proteins.
Color indicates the normalized Robinson–Foulds metric. The white arrow shows the junction of the two ORFs.
Рис. 3. Матрица филогенетической совместимости, построенная по объединенным выравниваниям белков NSP и SP.
Цветом показана нормализованная метрика Робинсона–Фолдса. Белой стрелкой показано место «стыка» двух рамок считывания.
Zoonotic risk assessment
N. Mollentze et al. [23] aimed to show a methodology to estimate the degree of human disease risk for viruses based on their genomic sequence. The machine learning model developed by the authors used phylogenetic information, nucleotide and dinucleotide composition of the viral genome, its similarity to interferon-stimulated genes, etc. The use of all these features made it possible to calculate an integral characterization reflecting the probability of zoonotic diseases. The application of this methodology on the two bastrovirus genomes obtained in this study demonstrated the following results: bastrovirus from sample 3 was categorized as High (i.e., having a high zoonotic potential) while the virus from sample 4 was categorized as Medium.
Discussion
Within the framework of the study, based on metagenomic sequencing of genetic material of bat fecal samples caught in the Russian Federation in 2023, nearly complete consensus bastrovirus genome sequences were successfully assembled in 2 samples of N. noctula species. Thus, in one of them, due to de novo assembly, a nearly complete (5832 bases) bastrovirus genome sequence was obtained, which allowed its use as a reference for genome assembly (5669 bases) in the other sample. Based on the results of genome annotation and comparative analysis of amino acid sequences, it was found that the viruses under consideration have significant differences compared to those already known, and their similarity to the closest homologs does not exceed 77 and 63% for the NSP protein and the SP protein respectively. Phylogenetic analysis confirmed their relative uniqueness and that the closest homologs are bastrovirus sequences from Vietnam. Genomic sequences were uploaded to the VGARus (crie051639, crie051640) and NCBI GenBank (OR552430, OR552431) databases.
Recombination is a frequent phenomenon in RNA viruses [26]. Our analysis using phylogenetic compatibility matrices showed the presence of a recombination event within the virus sequence, which should be taken into account when constructing phylogenies. Two clades of bat bastroviruses are distinguished and the topologies of trees constructed based on different parts of the genome do not coincide. All this indicates a continuous recombination process in bastroviruses, which can potentially lead to the rapid emergence of new properties of the virus, and given the high zoonotic potential of the virus, calculated by machine learning methods, further investigation is of utmost importance. Thus, the first detection of bastrovirus in the Russian Federation adds to the global data on the width of the bastrovirus distribution range, and the new data not only expand our understanding of the genetic diversity of bastroviruses, but also emphasize the importance of future studies to determine the potential impact of these viruses on human and animal health. Nucleotide sequences have been uploaded to the VGARus and NCBI GenBank databases.
Conclusion
In conclusion, the emergence and development of next-generation sequencing (NGS) technologies has not only revolutionized many areas of biology and medicine, but is also increasingly being used in virology. Thus, new approaches allow the study of a huge number of viruses, and as the cost of NGS [27] decreases, large-scale metagenomic studies, including for the detection of novel viral pathogens, are becoming increasingly affordable. The advantage of metagenomic sequencing is that it does not require the use of specific probes or primers for virus detection, potentially allowing the detection of any pathogen present in a sample, regardless of whether it is known or not [28]. This unique property has made metagenomic sequencing the primary method for detecting both known and novel viruses [29 30], which is particularly relevant for analyzing natural virus reservoirs such as bats . Thus, the ability to characterize multiple viruses in such sites, whether they are host-specific pathogens or considered as food or environmental samples, is a factor of importance and may contribute to a better understanding of some zoonotic transmission events and alert public health and epidemiologic surveillance authorities to their possible occurrence.
About the authors
German V. Roev
Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing; Moscow Institute of Physics and Technology (National Research University)
Email: roevherman@gmail.com
ORCID iD: 0000-0002-2353-5222
Bioinformatician of Laboratory for Genomics Research of the Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, Moscow, Russia
Russian Federation, 111123, Moscow; 115184, DolgoprudnyNadezhda I. Borisova
Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing
Email: borisova@cmd.su
ORCID iD: 0000-0002-9672-0648
Junior researcher of Laboratory for Genomics Research of the Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, Moscow, Russia
Russian Federation, 111123, MoscowNadezhda V. Chistyakova
A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences
Email: lanche@mail.ru
ORCID iD: 0009-0002-6034-1408
Engineer of Laboratory of comparative etology and biocommunication of A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences, Russia, Moscow, Russia
Russian Federation, 119071, MoscowAnastasia V. Vyhodtseva
Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing
Email: vihodceva@cmd.su
ORCID iD: 0009-0005-1911-9620
Technologist of Laboratory for Genomics Research of the Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, Moscow, Russia
Russian Federation, 111123, MoscowVasiliy G. Akimkin
Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
Doctor of Medicine, Professor, Director of Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, Moscow, Russia
Russian Federation, 111123, MoscowKamil F. Khafizov
Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing
Author for correspondence.
Email: khafizov@cmd.su
ORCID iD: 0000-0001-5524-0296
PhD (Biol.), Head of Laboratory for Genomics Research of the Central Research Institute for Epidemiology of the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing, Moscow, Russia
Russian Federation, 111123, MoscowReferences
- Oude Munnink B.B., Cotten M., Canuti M., Deijs M., Jebbink M.F., van Hemert F.J., et al. A Novel astrovirus-like RNA virus detected in human stool. Virus Evol. 2016; 2(1): vew005. https://doi.org/10.1093/ve/vew005
- Dos Anjos K., Nagata T., Melo F.L. Complete genome sequence of a novel bastrovirus isolated from raw sewage. Genome Announc. 2017; 5(40): e01010–17. https://doi.org/10.1128/genomeA.01010-17
- Yinda C.K., Ghogomu S.M., Conceição-Neto N., Beller L., Deboutte W., Vanhulle E., et al. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018; 4(1): vey008. https://doi.org/10.1093/ve/vey008
- Bauermann F.V., Hause B., Buysse A.R., Joshi L.R., Diel D.G. Identification and genetic characterization of a porcine hepe-astrovirus (bastrovirus) in the United States. Arch. Virol. 2019; 164(9): 2321–6. https://doi.org/10.1007/s00705-019-04313-x
- Mishra N., Fagbo S.F., Alagaili A.N., Nitido A., Williams S.H., Ng J., et al. A viral metagenomic survey identifies known and novel mammalian viruses in bats from Saudi Arabia. PLoS One. 2019; 14(4): e0214227. https://doi.org/10.1371/journal.pone.0214227
- Nagai M., Okabayashi T., Akagami M., Matsuu A., Fujimoto Y., Hashem M.A., et al. Metagenomic identification, sequencing, and genome analysis of porcine hepe-astroviruses (bastroviruses) in porcine feces in Japan. Infect. Genet. Evol. 2021; 88: 104664. https://doi.org/10.1016/j.meegid.2020.104664
- Chen Z., Zhao H., Li Z., Huang M., Si N., Zhao H., et al. First discovery of phenuiviruses within diverse RNA viromes of Asiatic toad (Bufo gargarizans) by metagenomics sequencing. Viruses. 2023; 15(3): 750. https://doi.org/10.3390/v15030750
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30(15): 2114–20. https://doi.org/10.1093/bioinformatics/btu170
- Bushnell B., Rood J., Singer E. BBMerge – accurate paired shotgun read merging via overlap. PLoS One. 2017; 12(10): e0185056. https://doi.org/10.1371/journal.pone.0185056
- Menzel P., Ng K.L., Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016; 7: 11257. https://doi.org/10.1038/ncomms11257
- Li D., Liu C.M., Luo R., Sadakane K., Lam T.W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015; 31(10): 1674–6. https://doi.org/10.1093/bioinformatics/btv033
- Buchfink B., Reuter K., Drost H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 2021; 18(4): 366–8. https://doi.org/10.1038/s41592-021-01101-x
- Langmead B., Wilks C., Antonescu V., Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019; 35(3): 421–32. https://doi.org/10.1093/bioinformatics/bty648
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021; 10(2): giab008. https://doi.org/10.1093/gigascience/giab008
- Wheeler D.L., Church D.M., Federhen S., Lash A.E., Madden T.L., Pontius J.U., et al. Database resources of the National Center for Biotechnology. Nucleic. Acids Res. 2003; 31(1): 28–33. https://doi.org/10.1093/nar/gkg033
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990; 215(3): 403–10. https://doi.org/10.1016/S0022-2836(05)80360-2
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30(4): 772–80. https://doi.org/10.1093/molbev/mst010
- Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015; 32(1): 268–74. https://doi.org/10.1093/molbev/msu300
- Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14(6): 587–9. https://doi.org/10.1038/nmeth.4285
- Letunic I., Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. Acids Res. 2021; 49(W1): W293–6. https://doi.org/10.1093/nar/gkab301
- Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006; 34(Web Server issue): W609–12. https://doi.org/10.1093/nar/gkl315
- Martin D.P., Murrell B., Golden M., Khoosal A., Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015; 1(1): vev003. https://doi.org/10.1093/ve/vev003
- Mollentze N., Babayan S.A., Streicker D.G. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 2021; 19(9): e3001390. https://doi.org/10.1371/journal.pbio.3001390
- Wolfaardt M., Kiulia N.M., Mwenda J.M., Taylor M.B. Evidence of a recombinant wild-type human astrovirus strain from a Kenyan child with gastroenteritis. J. Clin. Microbiol. 2011; 49(2): 728–31. https://doi.org/10.1128/JCM.01093-10
- Wohlgemuth N., Honce R., Schultz-Cherry S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019; 69: 30–7. https://doi.org/10.1016/j.meegid.2019.01.009
- Worobey M., Holmes E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999; 80(Pt. 10): 2535–43. https://doi.org/10.1099/0022-1317-80-10-2535
- van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014; 30(9): 418–26. https://doi.org/10.1016/j.tig.2014.07.001
- Kiselev D., Matsvay A., Abramov I., Dedkov V., Shipulin G., Khafizov K. Current trends in diagnostics of viral infections of unknown etiology. Viruses. 2020; 12(2): 211. https://doi.org/10.3390/v12020211
- Radford A.D., Chapman D., Dixon L., Chantrey J., Darby A.C., Hall N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012; 93(Pt. 9): 1853–68. https://doi.org/10.1099/vir.0.043182-0
- Bassi C., Guerriero P., Pierantoni M., Callegari E., Sabbioni S. Novel virus identification through metagenomics: a systematic review. Life (Basel). 2022; 12(12): 2048. https://doi.org/10.3390/life12122048
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005; 310(5748): 676–9. https://doi.org/10.1126/science.1118391
Supplementary files

