



Бастровирусы (Astroviridae): генетическое разнообразие и потенциальное влияние на здоровье человека и животных
- Авторы: Роев Г.В.1,2, Борисова Н.И.1, Чистякова Н.В.3, Выходцева А.В.1, Акимкин В.Г.1, Хафизов К.Ф.1
-
Учреждения:
- ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
- ФГАОУ ВО «Московский физико-технический институт (национальный исследовательский университет)»
- ФБУН «Институт проблем экологии и эволюции им. А.Н. Северцова» РАН
- Выпуск: Том 68, № 6 (2023)
- Страницы: 505-512
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://virusjour.crie.ru/jour/article/view/14736
- DOI: https://doi.org/10.36233/0507-4088-192
- EDN: https://elibrary.ru/iqblwy
- ID: 14736

Цитировать

Аннотация
Введение. Бастровирусы были обнаружены в Нидерландах в 2016 г. в кале человека. Они демонстрируют частичное генетическое сходство с астровирусами, а также вирусами гепатита Е. Их связь с возникновением заболеваний пока не была установлена.
Цель работы. Получение новой генетической информации о бастровирусах, циркулирующих среди летучих мышей на территории России.
Материалы и методы. Было проведено метагеномное секвенирование образцов фекалий летучих мышей вида Nyctalus noctula, выловленных на территории РФ в 2023 г. Собрано два почти полных генома бастровирусов. Был оценен зоонозный потенциал данных вирусов методами машинного обучения, изучена их рекомбинация, а также построены филогенетические деревья.
Результаты. De novo был собран почти полный геном (длина около 5800 оснований) нового бастровируса в одном из образцов, он был использован как референс для получения другого генома в другом образце. Зоонозный потенциал вируса одного из этих образцов методами машинного обучения был оценен как высокий. Показано существование рекомбинации между структурным и неструктурным полипротеинами.
Заключение. Первое обнаружение бастровируса на территории РФ дополняет мировые данные о широте ареала его распространения. Наличие рекомбинации между полипротеинами и высокий зоонозный потенциал вируса подчеркивают важность его дальнейшего изучения.
Ключевые слова
Полный текст
Введение
В 2016 г. в Нидерландах при проведении метагеномного секвенирования 200 образцов кала людей был найден ранее неизвестный РНК-вирус [1]. В 7 из этих образцов был идентифицирован новый патоген, имеющий некоторое генетическое сходство с астровирусом и вирусом гепатита Е. Длина последовательностей варьировала от 6017 до 6339 оснований. Вирус во всех 7 образцах имел одну и ту же структуру генома, а сходство аминокислотных последовательностей предполагаемого неструктурного белка ORF1 варьировало от 67 до 93%. Аналогично, сходство потенциального структурного белка ORF2 составляло от 73 до 98%. Такая высокая степень разнообразия позволила авторам статьи сделать предположение, что бастровирус как инфекционный агент человека, скорее всего, циркулировал среди человечества в течение длительного времени, а не появился недавно. Подобное разнообразие также может быть связано с возникновением в разных переносчиках инфекции и передачей человеку через загрязненную пищу или в качестве зоонозной инфекции от домашних животных, скота или диких животных. Все геномы бастровирусов содержали схожие консервативные аминокислотные домены. Капсидный белок бастровирусов демонстрирует заметную вариабельность, особенно в начальной 40-нуклеотидной N-концевой и конечной 242-нуклеотидной C-концевой последовательностях. На С-конце капсидного белка обнаружено несколько антигенных эпитопов, имеющих длину более десяти аминокислот. Проведение вложенной полимеразной цепной реакции (ПЦР), направленной на выявление 5’-области генома, позволило идентифицировать бастровирус в 32 из 200 исследованных образцов фекалий. Однако явной корреляции между клиническим симптомом, таким как диарея, и наличием вируса выявлено не было [1].
С момента первоначального обнаружения бастровируса в фекалиях человека в 2016 г. его неоднократно определяли в различных образцах. В 2017 г. в сточных водах Бразилии был идентифицирован бастровирус c размером генома 5875 оснований. Идентичность его генома составила 56% по сравнению с ранее известными бастровирусами (GenBank: KX907135) [2]. Исследователи предположили, что его переносчиком может быть млекопитающее. В исследовании 2018 г. бастровирус (GenBank: MG693175) был идентифицирован в 87 образцах фекалий летучих мышей двух разных видов в Камеруне [3]. В исследовании 2019 г. во время вспышки диареи свиней в США [4] в образцах фекалий был обнаружен геном бастровируса длиной 5881 основание с 84% идентичностью бастровирусу свиней (GenBank: KX907134), который впоследствии получил название PBastV-USA 2017-1. Анализ последовательности его генома выявил 97 и 87% сходства с последовательностями ORF1 и ORF2 обнаруженного бастровируса соответственно, что свидетельствует о значительном генетическом сходстве с бастровирусом свиней. Эти данные о последовательности полного генома позволили создать тест на основе количественной ПЦР для выявления патогена. В июне 2017 г. этот тест был использован для изучения 368 образцов свиней, поступивших в лабораторию болезней животных Университета штата Южная Дакота. Основную часть из них составляли образцы мазков из ротовой полости (90%), а остальные (10%) – фекальные, ректальные мазки или образцы из окружающей среды. Из 368 исследованных образцов 114 (или 31%) оказались положительными на вирус. При этом следует отметить, что большинство образцов были получены от здоровых свиней, так как были взяты в рамках инициативы по надзору за заболеваниями животных на фермах. Таким образом, обнаруженный бастровирус не был связан с каким-либо конкретным заболеванием.
В 2019 г. вышла статья, посвященная изучению 72 образцов летучих мышей четырех видов, выловленных в 2012 г. в городе Биша и его окрестностях в Саудовской Аравии [5]. У двух видов летучих мышей нашли вирус, схожий с бастровирусом, но обнаруженные вирусы в Саудовской Аравии были ближе к гепевирусам, чем к человеческому бастровирусу, имея 50% сходство по аминокислотной последовательности с ним. В результате исследователи классифицировали их как Middle East Hepe-Astrovirus, так как они демонстрировали около 70% сходства с бастровирусом летучих мышей и бастровирусом крысы из Вьетнама.
В 2021 г. японские исследователи идентифицировали практически полные последовательности геномов четырех бастровирусов, обнаруженных в фекалиях здоровых свиней [6]. Они обнаружили, что бастровирусы, присутствующие в организме свиней и других животных, включая человека, имеют схожую геномную организацию. В частности, все они имеют три консервативных домена: вирусную метилтрансферазу, РНК-хеликаз и RdRp в неструктурном ORF1 и капсидный домен астровируса в структурном ORF2. Сравнение аминокислотных последовательностей показало, что полученные бастровирусы имеют 95–99 и 76–96% сходства в областях ORF1 и ORF2 соответственно. Однако при сравнении бастровирусов свиней с бастровирусами других животных сходство составило лишь 21–43 и 9–21% для областей ORF1 и ORF2 соответственно. Это указывает на то, что хотя бастровирусы могли иметь общего предка, они эволюционировали отдельно в каждой группе хозяев в течение длительного времени. Наличие в геноме потенциальных рекомбинационных событий также свидетельствует о том, что бастровирусы приобретают генетическое разнообразие в результате рекомбинационных событий.
В 2023 г. была опубликована работа [7], посвященная малоизученному вирусному разнообразию низших позвоночных, включая рыб, земноводных и рептилий. Объектом изучения служила азиатская жаба, обитающая в Китае. В результате проведенных исследований было выявлено более 20 новых РНК-вирусов в материалах жаб. Так, выявлен практически полный геном бастровируса, сильно отличающийся от ранее известных вариантов, что позволило выделить новые ветви на филогенетическом дереве. Этот геном, обозначенный как AtBastV/GCCDC11/2022, состоит из трех предполагаемых белок-кодирующих областей, каждая из которых имеет разную степень идентичности с различными известными вирусами. При более глубоком изучении филогении RdRp и капсидных сегментов AtBastV/GCCDC11 было обнаружено, что он имеет сильное генетическое сходство со штаммом бастровируса амфибий из рода Rana. Кроме того, этот штамм имел заметное сходство с астровирусом 2, обнаруженным у хайнаньской черноглазой жабы, и некоторыми астровирусами животных, хотя и имел отдаленное отношение к гепевирусам.
Филогенетический анализ, проведенный на основе последовательности РНК-зависимой РНК-полимеразы (RdRP), показывает, что бастровирусы и вирусы гепатита Е формируют на дереве отдельную кладу, отличную от астровирусов [3]. Большинство бастровирусов животных образуют монофилетическую группу и группируются по видам хозяев, которых они заражают. Исключением является линия Bat_Bastrovirus-like_virus/VietNam/Bat/17819_21, образующая на дереве отдельную внешнюю группу. Филогенетический анализ по белку капсида дает схожие результаты. В статье 2016 г. [1] была высказана гипотеза, о том что между участками генома, кодирующими белок капсида и полимеразу, в прошлом произошла рекомбинация. В качестве свидетельства авторы указывают на то, что филогенетически белок капсида больше связан с астровирусами, а ген полимеразы – с вирусом гепатита Е. На дополнительные события рекомбинации также указывает тот факт, что штамм CMR/Bat/P24 группируется с двумя штаммами летучих мышей на дереве полимеразы и только с одним штаммом на дереве белка капсида [3].
Бастровирусы можно дифференцировать на основе генетических признаков. ORF1 всех бастровирусов млекопитающих кодирует вирусную метилтрансферазу, за которой следуют вирусная геликаза и RdRp, тогда как ORF2 кодирует капсидный белок. ORF3 присутствует в геноме не всех бастровирусов, а если и присутствует, как в случае бастровирусов человека, то кодирует папаин-подобную цистеиновую протеазу вируса гепатита Е. Хотя астровирусы и бастровирусы имеют некоторое сходство в структурном белке, структура генома бастровирусов и его особенности могут способствовать будущей классификации этих вирусов в новое вирусное семейство. Кроме того, обширное генетическое разнообразие среди бастровирусов, полученных от разных видов хозяев, может потребовать дальнейшей классификации бастровирусов в разные линии или роды.
Рассмотренные выше источники позволяют сделать вывод об очень широком распространении бастровирусов среди человека и различных видов животных. Бастровирусы выявлены у человека, свиней, летучих мышей, речных моллюсков и жаб и даже в сточных водах. Такое разнообразие носителей вируса подтверждает теорию о том, что данный вирус циркулирует в природе уже в течение какого-то периода времени, а не зародился недавно. До сих пор в литературе не было описано случаев обнаружения бастровирусов на территории России. В настоящей работе мы провели метагеномное секвенирование генетического материала образцов фекалий летучих мышей вида Nyctalus noctula из Саратовской области, в результате чего был обнаружен неизвестный ранее бастровирус и собран его полный геном. Мы провели аннотирование нового генома бастровируса, построили филогенетические деревья и провели рекомбинационный анализ.
Материалы и методы
Проведено метагеномное секвенирование образцов фекалий летучих мышей вида N. noctula, собранных в феврале 2023 г. в России. Выделение РНК из образцов выполняли с использованием набора QIAamp Viral RNA (Qiagen, Германия). Обратную транскрипцию осуществляли с использованием набора РЕВЕРТА-L («АмплиСенс», Россия) в соответствии с инструкцией производителя. Синтез второй цепи ДНК был выполнен с помощью набора NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module (E6111L). Очищенную двухцепочечную кДНК фрагментировали в microTUBE-50 AFA Fiber Screw-Cap (PN 520166) на приборе Covaris M220 (Covaris, Woburn, США) до ~ 550 п.о. Библиотеки для парноконцевого секвенирования сконструировали с помощью наборов NEBNext Ultra End Repair/dA-Tailing Module (NEB E7546L), NEBNext Ultra Ligation Module (NEB E7595L) и Y-адаптера, совместимого с IDT for Illumina Nextera DNA UD Indexes. Индексирующую ПЦР проводили с использованием NEBNext Ultra II Q5 Master Mix (NEB #M0492). Финальную библиотеку валидировали на приборе Agilent Bioanalyzer 2100 (Agilent Technologies, США). Секвенирование проводили на платформах MiSeq и NextSeq 2000 (Illumina, США).
Сборка консенсусных последовательностей
В «сырых» прочтениях удаляли адаптеры при помощи опции в Trimmomatic v0.39 ILLUMINACLIP [8]. Также применяли опции LEADING:7, TRAILING:7, SLIDINGWINDOW:4:20, и MINLEN:40. Парные прочтения объединяли при помощи BBmerge [9] (maxstrict=t), файлы с непарными прочтениями для каждого образца объединяли в один. Таким образом, для каждого образца получили по три файла: два файла – парно-концевые чтения и один файл – непарные. Прочтения, соответствующие хосту, удаляли выравниванием на референсные геномы летучих мышей Pipistrellus kuhlii (GCF_014108245.1) и Myotis myotis (GCF_014108235.1) при помощи bowtie2 (опции –un и –un-conc отдельно соответственно для непарных и парных ридов). Использовали геномы этих двух видов, так как референсный геном для N. noctula отсутствует в доступных базах данных. Далее с использованием Kaiju v1.9.2 [10] (использовалась база nr_euk) оставили только прочтения либо определяющиеся как вирусные, либо не классифицирующиеся (файлы с парно-концевыми чтениями и непарными снова обрабатывали отдельно). Оставшиеся после такой фильтрации прочтения были собрали de novo в контиги с использованием MEGAHIT v1.2.9 [11], получившиеся контиги использовали для поиска в базе NCBI nr при помощи DIAMOND [12] blastx (опции -very-sensitive, evalue 1e-08, -k 3).
В образце N.noctula_3 (MiSeq) на основе поиска по гомологии был найден длинный (5,832 bp) контиг, относящийся к бастровирусам. Этот контиг в дальнейшем использовали как референс для сборки консенсусов для другого образца. Также контиги бастровируса обнаружили в образцах фекалий летучих мышей N.noctula_4 (NextSeq), N.noctula_4 (MiSeq, техническая реплика предыдущего образца). Фильтрованные при помощи Kaiju прочтения с соответствующих образцов выравнивали bowtie2 v2.4.4 [13] (--local) на вышеуказанный референс. «Bam»-файлы реплик объединяли командой «samtools merge» [14] v1.15.1, сборку консенсусов производили командой «samtools consensus» (-m simple -aa -c 0.51). В результате получили две консенсусных последовательности (образцы № 3, 4).
Филогенетический анализ
В собранных последовательностях были выделены открытые рамки считывания с использованием NCBI ORFfinder [15] (опция «ATG» and alternative initiation codons) и BLASTX [16]. Аминокислотные последовательности, соответствующие неструктурному полипротеину (NSP) и структурному полипротеину (SP), использовали для дальнейшего филогенетического анализа. Для построения деревьев использовали 31 последовательность генома бастровирусов из NCBI, у которых были проаннотированы полипротеины NSP и SP. Список accession numbers использовавшихся образцов: KU318321.1, NC_035758.1, KU318317.1, KU318320.1, KU318315.1, KU318318.1, KU318319.1, KU318316.1, KX907134.1, NC_032423.1, MK387176.1, KX907130.1, MT549856.1, LC549662.1, MF042208.1, KX907133.1, NC_035471.1, OM104033.1, OQ835729.1, KX907129.1, KX907131.1, NC_032484.1, KX907132.1, KX907128.1, KX907127.1, MG693175.1, MT549857.1, KX907135.1, NC_032426.1, OQ835730.1, MT766313.1. Последовательность астровируса гуся (GenBank: OM200916.1) использовали как аутгруппу. Выравнивание осуществляли с помощью программы MAFFT v7.490 [17]. Построение дерева производили в IQ-TREE v2.2.3 [18] (опция -alrt 1000), оптимальную модель определяли при помощи ModelFinder [19]. Визуализацию дерева выполняли с применением онлайн-инструмента iTOL [20].
Анализ рекомбинации
Для изучения потенциальной рекомбинации в программе MAFTT были построены множественные выравнивания по аминокислотным последовательностям белков NSP и SP для 33 образцов (31 – получены из NCBI и 2 – в этом исследовании), которые затем обратно транслировали в нуклеотидные выравнивания при помощи PAL2NAL [21]. Далее выравнивания для белков NSP и SP объединяли. Матрицу филогенетической совместимости с метрикой Робинсона–Фолдса строили с применением протокола RDP4. Ширина окна составляла 400, шаг – 50 нуклеотидов [22].
Анализ зоонозного потенциала
Для оценки зоонозного потенциала бастровируса использовали обученную модель машинного обучения [23], доступную по ссылке https://github.com/Nardus/zoonotic_rank. На вход подавали два генома бастровируса, а также таблицу с аннотацией. С помощью команды PredictNovel.R получили файлы с результатами.
Авторы подтверждают соблюдение институциональных и национальных стандартов по использованию лабораторных животных в соответствии с Consensus author guidelines for animal use (IAVES 23 July 2010). Утверждено заключение этического комитета по работе с животными, номер 50 от 07.08.2021. Все процедуры проводились в соответствии с разрешением комиссии по биоэтике ИПЭЭ РАН им. А.Н. Северцова.
Результаты
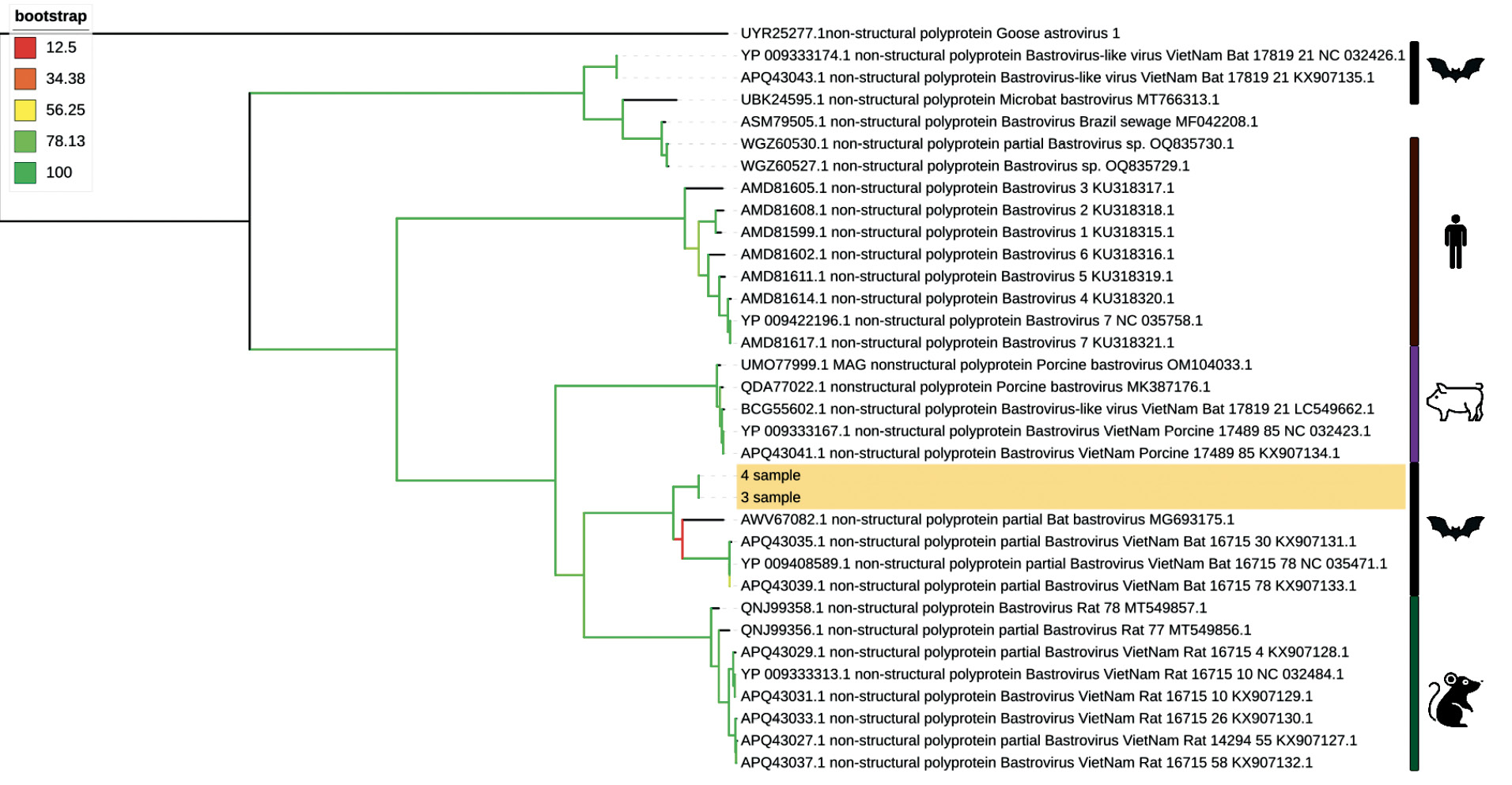
В настоящем исследовании на основе данных метагеномного секвенирования генетического материала фекалий летучих мышей N. noctula, пойманных на территории Российской Федерации в 2023 г., были получены практически полные последовательности генома бастровируса для двух образцов. В одном из образцов был собран de novo длинный контиг (около 5800 п.о.), покрывающий практически полный геном бастровируса, который затем использовали как референс для сборки генома бастровируса из другого образца. После аннотации геномов путем поиска с помощью программ BLASTX в базе NCBI nr и инструмента NCBI ORFfinder для этих двух образцов удалось получить аминокислотную последовательность NSP длиной около 1200 аминокислот и SP длиной порядка 630 аминокислот. Филогенетический анализ, проведенный по неструктурному (рис. 1) и структурному (рис. 2) полипротеинам показал сходство с образцами бастровируса из летучих мышей, пойманных во Вьетнаме. По результатам, полученным с помощью программы BLASTP, сходство с уже известными последовательностями по белку NSP составляет менее 77%, а по SP белку – менее 63%, что дает основание полагать, что вирусы из данных образцов являются новыми.

Рис. 1. Филогенетическое дерево, построенное на основе аминокислотной последовательности неструктурного полипротеина (NSP) бастровирусов.
Образцы, отсеквенированные в рамках этого исследования, выделены желтым. Цветом показана поддержка узлов, рассчитанная по методу SH-aLRT.
Fig. 1. Phylogenetic tree of the non-structural polyprotein (NSP) of bastroviruses.
Samples sequenced as part of this study are highlighted in yellow. Branch colors indicate node support calculated by the SH-aLRT method.

Рис. 2. Филогенетическое дерево, построенное на основе аминокислотной последовательности структурного полипротеина (SP) бастровирусов.
Образцы, отсеквенированные в рамках этого исследования, выделены желтым. Цветом показана поддержка узлов, рассчитанная по методу SH-aLRT.
Fig. 2. Phylogenetic tree of the structural polyprotein (SP) of bastroviruses.
Samples sequenced as part of this study are highlighted in yellow. Branch colors indicate node support calculated by the SH-aLRT method.
Изучение рекомбинации в геномах бастровирусов
Анализ последовательностей геномов бастровирусов с помощью матрицы филогенетической совместимости (рис. 3) указывает на наличие возможной рекомбинации между рамками считывания, кодирующими полипротеины SP и NSP, что в целом типично для астровирусов [24, 25], ближайшими родственниками которых являются бастровирусы. Также это подтверждается разной топологией филогенетических деревьев, построенных по белкам SP и NSP. Так, например, образец KX907131.1, представленный на рис. 1, находится в одной кладе с полученными в настоящем исследовании образцами, а на рис. 2 он показан в другой кладе бастровирусов летучих мышей.

Рис. 3. Матрица филогенетической совместимости, построенная по объединенным выравниваниям белков NSP и SP.
Цветом показана нормализованная метрика Робинсона–Фолдса. Белой стрелкой показано место «стыка» двух рамок считывания.
Fig. 3. Phylogenetic compatibility matrix constructed from the merged alignments of NSP and SP proteins.
Color indicates the normalized Robinson–Foulds metric. The white arrow shows the junction of the two ORFs.
Оценка степени зоонозного риска
N. Mollentze и соавт. [23] была предложена методология оценки степени риска заболевания человеком для вирусов, исходя из их геномной последовательности. Обученная авторами модель машинного обучения использовала филогенетическую информацию, нуклеотидный и динуклеотидный состав вирусного генома, его сходство с интерферон-стимулированными генами и т.д. Использование всех этих признаков позволило вычислить интегральную характеристику, отражающую вероятность зоонозов. Применение этой методологии на двух геномах бастровируса, полученных в настоящем исследовании, продемонстрировало следующие результаты: бастровирус из 3-го образца был отнесен к категории High (т.е. имеющий высокий зоонозный потенциал), из 4-го образца – Medium (средний).
Обсуждение
В рамках проведенного исследования на основе метагеномного секвенирования генетического материала образцов фекалий летучих мышей, пойманных в Российской Федерации в 2023 г., были успешно собраны практически полные консенсусные последовательности генома бастровируса в 2 образцах вида N. noctula. Так, в одном из них, благодаря de novo сборке, получена практически полная (5832 оснований) последовательность генома бастровируса, что позволило использовать ее в качестве референса для сборки генома (5669 оснований) в другом образце. По результатам аннотации геномов и сравнительного анализа аминокислотных последовательностей, установлено, что рассматриваемые вирусы обладают значительными различиями по сравнению с уже известными, и их сходство с ближайшими гомологами не превышает 77% по белку NSP и 63% по белку SP. Филогенетический анализ подтвердил их относительную уникальность и выявил их ближайшие гомологи – образцы бастровирусов из Вьетнама. Геномные последовательности были загружены в базы данных VGARus (crie051639, crie051640) и NCBI GenBank (OR552430, OR552431).
Рекомбинация – частое явление у РНК-вирусов [26]. Проведенный нами анализ с помощью матриц филогенетической совместимости показал наличие события рекомбинации внутри вируса, что должно учитываться при построении филогений. Выделяются две клады бастровирусов летучих мышей, причем топологии деревьев, построенных по разным участкам генома, не совпадают. Все это указывает на непрерывно идущий процесс рекомбинации у бастровирусов, что потенциально может быстро привести к появлению у вируса новых свойств, а учитывая высокий зоонозный потенциал вируса, вычисленный методами машинного обучения, важно его дальнейшее изучение. Таким образом, первое обнаружение бастровируса на территории РФ дополняет мировые данные о широте ареала распространения бастровируса, а новые данные не только расширяют наше понимание генетического разнообразия бастровирусов, но и подчеркивают важность будущих исследований для определения потенциального влияния этих вирусов на здоровье человека и животных. Нуклеотидные последовательности были загружены в базы данных VGARus и NCBI GenBank.
Заключение
Появление и развитие технологий секвенирования нового поколения (NGS) не только произвело революцию во многих областях биологии и медицины, но и все чаще используется в вирусологии. Так, новые подходы позволяют исследовать огромное количество вирусов, и по мере снижения стоимости NGS [27] масштабные метагеномные исследования, в том числе для выявления новых вирусных патогенов, становятся все более доступными. Преимуществом метагеномного секвенирования является отсутствие необходимости использования специфических зондов или праймеров для выявления вирусов, что потенциально позволяет обнаружить любой патоген, присутствующий в образце, независимо от его известного или неизвестного статуса [28]. Благодаря этому уникальному свойству метагеномное секвенирование стало основным методом обнаружения как известных, так и новых вирусов [29, 30], что особенно актуально для анализа таких природных резервуаров вирусов, как летучие мыши [31]. Возможность описывать множество вирусов в таких объектах, независимо от того являются ли они патогенами, специфичными для хозяина, или рассматриваются в качестве пищевых или экологических образцов, может способствовать лучшему пониманию некоторых зоонозных событий передачи инфекции и предупредить органы здравоохранения и эпидемиологического надзора о возможном их возникновении.
Об авторах
Герман Викторович Роев
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора; ФГАОУ ВО «Московский физико-технический институт (национальный исследовательский университет)»
Email: roevherman@gmail.com
ORCID iD: 0000-0002-2353-5222
биоинформатик лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Москва, Россия
Россия, 111123, г. Москва; 115184, г. ДолгопрудныйНадежда Ивановна Борисова
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: borisova@cmd.su
ORCID iD: 0000-0002-9672-0648
младший научный сотрудник лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Москва, Россия
Россия, 111123, г. МоскваНадежда Владимировна Чистякова
ФБУН «Институт проблем экологии и эволюции им. А.Н. Северцова» РАН
Email: lanche@mail.ru
ORCID iD: 0009-0002-6034-1408
инженер лаборатории сравнительной этологии и биокоммуникации ИПЭЭ РАН им. А.Н. Северцова, Москва, Россия
Россия, 119071, г. МоскваАнастасия Владимировна Выходцева
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: vihodceva@cmd.su
ORCID iD: 0009-0005-1911-9620
технолог лаборатории геномных исследований ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Москва, Россия
Россия, 111123, г. МоскваВасилий Геннадьевич Акимкин
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
д-р мед. наук, профессор, директор ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Москва, Россия
Россия, 111123, г. МоскваКамиль Фаридович Хафизов
ФБУН «Центральный научно-исследовательский институт эпидемиологии» Роспотребнадзора
Автор, ответственный за переписку.
Email: khafizov@cmd.su
ORCID iD: 0000-0001-5524-0296
канд. биол. наук, заведующий лабораторией геномных исследований, ФБУН ЦНИИ Эпидемиологии Роспотребнадзора, Москва, Россия
Россия, 111123, г. МоскваСписок литературы
- Oude Munnink B.B., Cotten M., Canuti M., Deijs M., Jebbink M.F., van Hemert F.J., et al. A Novel astrovirus-like RNA virus detected in human stool. Virus Evol. 2016; 2(1): vew005. https://doi.org/10.1093/ve/vew005
- Dos Anjos K., Nagata T., Melo F.L. Complete genome sequence of a novel bastrovirus isolated from raw sewage. Genome Announc. 2017; 5(40): e01010–17. https://doi.org/10.1128/genomeA.01010-17
- Yinda C.K., Ghogomu S.M., Conceição-Neto N., Beller L., Deboutte W., Vanhulle E., et al. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018; 4(1): vey008. https://doi.org/10.1093/ve/vey008
- Bauermann F.V., Hause B., Buysse A.R., Joshi L.R., Diel D.G. Identification and genetic characterization of a porcine hepe-astrovirus (bastrovirus) in the United States. Arch. Virol. 2019; 164(9): 2321–6. https://doi.org/10.1007/s00705-019-04313-x
- Mishra N., Fagbo S.F., Alagaili A.N., Nitido A., Williams S.H., Ng J., et al. A viral metagenomic survey identifies known and novel mammalian viruses in bats from Saudi Arabia. PLoS One. 2019; 14(4): e0214227. https://doi.org/10.1371/journal.pone.0214227
- Nagai M., Okabayashi T., Akagami M., Matsuu A., Fujimoto Y., Hashem M.A., et al. Metagenomic identification, sequencing, and genome analysis of porcine hepe-astroviruses (bastroviruses) in porcine feces in Japan. Infect. Genet. Evol. 2021; 88: 104664. https://doi.org/10.1016/j.meegid.2020.104664
- Chen Z., Zhao H., Li Z., Huang M., Si N., Zhao H., et al. First discovery of phenuiviruses within diverse RNA viromes of Asiatic toad (Bufo gargarizans) by metagenomics sequencing. Viruses. 2023; 15(3): 750. https://doi.org/10.3390/v15030750
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30(15): 2114–20. https://doi.org/10.1093/bioinformatics/btu170
- Bushnell B., Rood J., Singer E. BBMerge – accurate paired shotgun read merging via overlap. PLoS One. 2017; 12(10): e0185056. https://doi.org/10.1371/journal.pone.0185056
- Menzel P., Ng K.L., Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016; 7: 11257. https://doi.org/10.1038/ncomms11257
- Li D., Liu C.M., Luo R., Sadakane K., Lam T.W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015; 31(10): 1674–6. https://doi.org/10.1093/bioinformatics/btv033
- Buchfink B., Reuter K., Drost H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 2021; 18(4): 366–8. https://doi.org/10.1038/s41592-021-01101-x
- Langmead B., Wilks C., Antonescu V., Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019; 35(3): 421–32. https://doi.org/10.1093/bioinformatics/bty648
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021; 10(2): giab008. https://doi.org/10.1093/gigascience/giab008
- Wheeler D.L., Church D.M., Federhen S., Lash A.E., Madden T.L., Pontius J.U., et al. Database resources of the National Center for Biotechnology. Nucleic. Acids Res. 2003; 31(1): 28–33. https://doi.org/10.1093/nar/gkg033
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990; 215(3): 403–10. https://doi.org/10.1016/S0022-2836(05)80360-2
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30(4): 772–80. https://doi.org/10.1093/molbev/mst010
- Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015; 32(1): 268–74. https://doi.org/10.1093/molbev/msu300
- Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14(6): 587–9. https://doi.org/10.1038/nmeth.4285
- Letunic I., Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. Acids Res. 2021; 49(W1): W293–6. https://doi.org/10.1093/nar/gkab301
- Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006; 34(Web Server issue): W609–12. https://doi.org/10.1093/nar/gkl315
- Martin D.P., Murrell B., Golden M., Khoosal A., Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015; 1(1): vev003. https://doi.org/10.1093/ve/vev003
- Mollentze N., Babayan S.A., Streicker D.G. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 2021; 19(9): e3001390. https://doi.org/10.1371/journal.pbio.3001390
- Wolfaardt M., Kiulia N.M., Mwenda J.M., Taylor M.B. Evidence of a recombinant wild-type human astrovirus strain from a Kenyan child with gastroenteritis. J. Clin. Microbiol. 2011; 49(2): 728–31. https://doi.org/10.1128/JCM.01093-10
- Wohlgemuth N., Honce R., Schultz-Cherry S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019; 69: 30–7. https://doi.org/10.1016/j.meegid.2019.01.009
- Worobey M., Holmes E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999; 80(Pt. 10): 2535–43. https://doi.org/10.1099/0022-1317-80-10-2535
- van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014; 30(9): 418–26. https://doi.org/10.1016/j.tig.2014.07.001
- Kiselev D., Matsvay A., Abramov I., Dedkov V., Shipulin G., Khafizov K. Current trends in diagnostics of viral infections of unknown etiology. Viruses. 2020; 12(2): 211. https://doi.org/10.3390/v12020211
- Radford A.D., Chapman D., Dixon L., Chantrey J., Darby A.C., Hall N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012; 93(Pt. 9): 1853–68. https://doi.org/10.1099/vir.0.043182-0
- Bassi C., Guerriero P., Pierantoni M., Callegari E., Sabbioni S. Novel virus identification through metagenomics: a systematic review. Life (Basel). 2022; 12(12): 2048. https://doi.org/10.3390/life12122048
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005; 310(5748): 676–9. https://doi.org/10.1126/science.1118391
Дополнительные файлы

