



Endothelial activation and dysfunction caused by influenza A virus (Alphainfluenzavirus influenzae)
- Authors: Marchenko V.A.1, Zhilinskaya I.N.1
-
Affiliations:
- North-Western State Medical University Named after I.I. Mechnikov
- Issue: Vol 69, No 6 (2024)
- Pages: 465-478
- Section: REVIEWS
- URL: https://virusjour.crie.ru/jour/article/view/16681
- DOI: https://doi.org/10.36233/0507-4088-264
- EDN: https://elibrary.ru/zujoza
- ID: 16681

Cite item

Abstract
Annual epidemics of influenza result in 3–5 million cases of severe illness and more than 600 000 deaths. Severe forms of influenza are usually characterized by vascular endothelial cells damage. Thus, influenza A viruses, including subtypes A(H1N1)pdm09, A(H3N2), as well as highly pathogenic avian influenza viruses, can infect the vascular endothelium, leading to activation and subsequent dysfunction of these cells. In turn, endothelial dysfunction resulting in systemic morphofunctional changes of endothelial cells, which leads to impaired vascular tone, thrombosis and other complications, and is also a risk factor and profoundly implicated in the pathogenesis of many cardiovascular diseases. Thus, endothelial dysfunction is an important aspect of the pathogenesis of severe influenza, which must be considered in the pathogenetic therapy of this infectious disease.
The aim of the review is to analyze the causes and specify mechanisms of development of endothelial activation and dysfunction caused by influenza A virus.
Full Text
Introduction
Influenza viruses are one of the most common pathogens of respiratory infectious diseases. Thus, influenza affects more than 15% of the world’s population annually, while the severe course of influenza infection is registered in 3‒5 million patients [1]. In turn, the severe course of influenza often proceeds with the development of hemorrhagic syndrome, which increases the risk of hemorrhagic stroke, myocardial infarction, acute coronary syndrome, deep vein thrombosis and other cardiovascular complications [2‒5]. According to the World Health Organization, risk groups for the development of severe influenza include children under 6 years of age, people over 65 years of age, pregnant women, people with chronic somatic pathology and immunocompromised individuals.
The vascular endothelium is known to be a target for influenza type A viruses (IAVs) [6‒8]. In severe course of infection, influenza viruses mediate excessive activation and damage to the vascular endothelium, which causes the development of endothelial dysfunction (ED). In turn, ED is an important link in the pathogenesis, as well as a risk factor for the development of multiple cardiovascular pathologies.
This review presents current information concerning the mechanisms of activation and ED of blood vessels in IAV infection.
Functions of the vascular endothelium
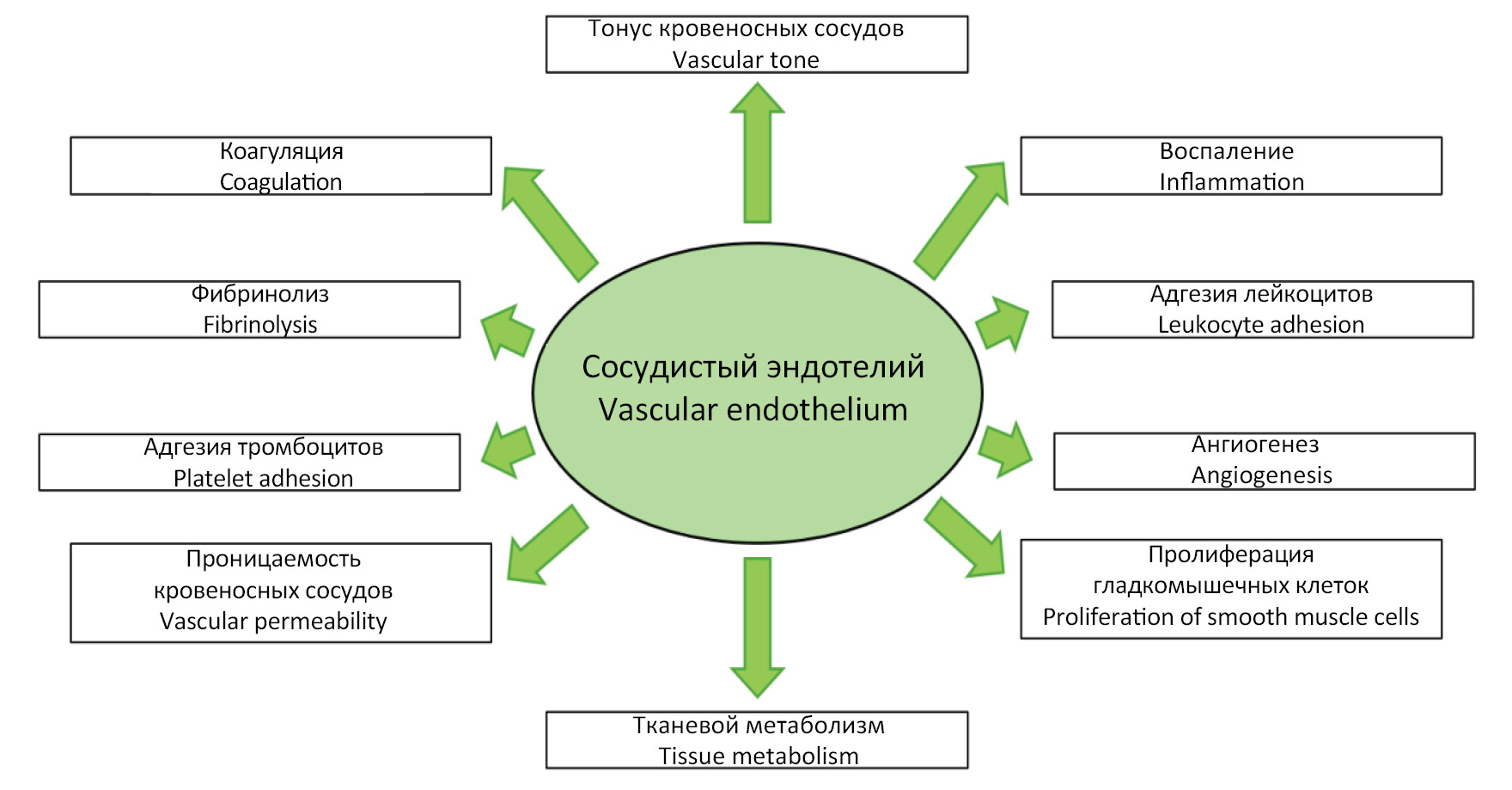
The endothelium is a giant endocrine organ distributed throughout all tissues of the human body, maintaining vascular and tissue homeostasis through the production of a variety of biologically active substances. Under physiologic conditions, anatomically and functionally intact vascular endothelium performs a number of functions, including: 1) regulation of blood vessel tone (vasomotor function); 2) regulation of leukocyte adhesion (adhesive function); 3) regulation of the hemostasis system (hemostatic function); 4) regulation of angiogenesis (angiogenic function); 5) regulation of immune processes (immune function), etc. (Fig. 1) [9, 10].

Fig. 1. Vascular endothelium functions.
Рис. 1. Функции эндотелия кровеносных сосудов.
Vascular endothelium as a target for influenza A viruses
IAVs are capable of infecting various cells of the respiratory tract, including cells of the mesenteric epithelium, as well as bronchiolar exocrinocytes [11, 12]. Influenza viruses utilize sialic acids as a receptor for adsorption. IAV subtypes A(H1N1)pdm09 and A(H3N2) are characterized by high specificity for the α-2,6-linked sialic acid, which is predominantly expressed on the epithelium of the upper and lower respiratory tracts, including tracheal and bronchial epithelium, as well as on type 1 alveolocytes [13‒15]. During intense reproduction, seasonal influenza viruses exert direct cytopathic effects on infected epitheliocytes, resulting in significant damage and death [16, 17]. This allows viruses to penetrate from the entry gate into regional blood vessels and interact with the vascular endothelium.
In turn, highly pathogenic avian influenza viruses specifically bind to α-2,3-sialic acid on the surface of lower respiratory tract epithelial cells, including bronchiolar epithelium and type 2 alveolocytes [18, 19]. Influenza virus replication in alveolocytes typically leads to cell apoptosis, allowing viruses to contact the basolateral surface of pulmonary capillary endothelial cells [20, 21].
Endothelial cells of blood vessels, including lung microvessels, have been found to express both types of sialic acids (α-2,3 and α-2,6) on their surface [14, 22, 23]. Thus, in vitro studies have shown that pulmonary capillary endothelial cells are sensitive to IAVs, but the titer of highly pathogenic influenza A(H5N1) and A(H7N9) viruses in endothelial cell culture is several orders of magnitude higher than the titer of IAV subtypes A(H1N1)pdm09 and A(H3N2) – 5‒8 lg vs. 2.5‒4 lg TCD50/mL [24‒26]. In vivo experiments also support the fact that IAVs can infect vascular endothelial cells and that highly pathogenic avian influenza viruses are much more likely to infect the vascular endothelium, leading to the development of life-threatening complications such as cytokine storm, acute respiratory distress syndrome (ARDS) and disseminated intravascular coagulation syndrome (DIC) [27‒29]. Thus, endothelial cells are not only sensitive but also permissive to highly pathogenic avian influenza viruses and influenza A(H1N1) and A(H3N2) viruses.
Endothelial activation and dysfunction
Endothelial activation and dysfunction are close but not identical concepts [30]. Thus, endothelial activation should be considered as a variant of cellular response to various stimuli, the intensity and/or duration of which does not exceed the limit of cellular adaptive response. Such activating stimuli include blood flow disturbance, cytokinemia, hypoxia, toxic substances, pathogen-associated molecular patterns (PAMP), damage-associated molecular patterns (DAMP), etc. [31‒33]. Furthermore, endothelial activation is observed when cells are exposed to reactive oxygen species (ROS) and reactive nitrogen species (RNS). On the one hand, an increase in the level of ROS is a necessary condition for the realization of innate immunity mechanisms, and on the other hand, in case of failure of the antioxidant defense system, it causes oxidative stress and damage to cell membranes [34].
Endothelial activation is a two-stage process. Thus, during the first stage (type I activation, or endothelial stimulation), endothelial cells respond almost immediately to a stimulus without a change in phenotype and de novo protein synthesis [35]. The interaction of ligand (e.g., histamine) with G-protein-coupled receptors mediates actin cytoskeleton rearrangement, cell contraction, and exocytosis of a number of proteins, including von Willebrand factor (vWF) and P-selectin, from Weibel‒Palade bodies to the cell surface [36].
The second stage (type II activation) occurs as a delayed response of endothelial cells when exposed to a stimulus over several hours or days [30]. Type II endothelial activation is based on cytokine-mediated activation of the NF-κB signaling pathway, resulting in increased expression of dozens of genes and a change in the phenotype of endothelial cells to a pro-inflammatory one [37, 38].
Endothelial activation can be either reversible or irreversible. If the effect of a trigger factor on the endothelium is limited in time, the genes supporting the pro-inflammatory phenotype are gradually inhibited, and the expression of genes of the vasoprotective phenotype is restored [31]. In turn, in case of overexpressed and/or prolonged endothelial activation, receptor, biochemical, and morphostructural changes are observed, which lead to endothelial cell damage and development of ED [39].
ED is characterized by persistent disturbance of morphofunctional characteristics of endothelial cells. In this pathological process, there is a dysregulation of vasodilation and vasoconstriction, coagulation and fibrinolysis, angiogenesis, inflammation and immune response. It is ED that is both a risk factor and the main link in the pathogenesis of many diseases of the cardiovascular system, including atherosclerosis, arterial hypertension, ischemic stroke and other pathologies [30, 40, 41]. Thus, influenza can initiate the development or aggravate the course of diseases of the cardiovascular system.
Activation and dysfunction of endothelial cells in influenza A: mechanisms
To date, numerous in vitro and in vivo studies have confirmed the fact that IAVs can cause not only activation but also dysfunction of blood vessel endothelial cells due to dysregulation of numerous cellular processes as a result of changes in the expression of more than 100 target genes [42]. The mechanisms of ED development in influenza are presented below.
Oxidative stress and decreased nitric oxide (NO) bioavailability
The cornerstone of endothelial dysfunction of blood vessels is considered to be a decrease in the synthesis and/or bioavailability of NO as a result of dysregulation of expression and/or activity of endothelial nitric oxide synthase (eNOS) [43]. Under physiologic conditions, eNOS is continuously expressed in the vascular endothelium and generates NO at low concentrations. This is extremely important because it is in low concentration that NO has anti-inflammatory, anti-proliferative, anti-thrombogenic and vasodilation effects [31].
When endothelial cells are infected with IAV, there is a change in the expression of this endothelial factor. Thus, influenza virus A(H1N1)pdm09 significantly reduces the level of eNOS expression in endothelial cell culture, and also causes a long-term and systemic decrease in the expression of this enzyme in vivo (at least 2 months) [8, 44]. These results are consistent with epidemiologic data on the positive correlation between influenza morbidity and mortality from cardiovascular diseases within 2 months after the end of the epidemic (also known as excess mortality) [45].
It has been established that one of the main causes of IAV-induced dysregulation of eNOS expression is free-radical damage of cells as a result of oxidative stress. When endothelium is activated, excessive formation of ROS by mitochondria occurs in cells. In case of antioxidant system failure, free radicals, in particular superoxide anion radical (O2−), are able to bind to NO with the formation of peroxynitrite (ONOO-), which has an extremely high oxidative potential [46]. In turn, peroxynitrite causes oxidation of tetrohydrobiopterin, one of the cofactors of eNOS, leading to dissociation of eNOS from its substrate, as a result of which this enzyme starts to produce superoxide anion radical instead of NO [47]. Thus, the concentration of ROS and RNS in the cell increases dramatically, which increases endothelial cell damage and aggravates ED.
It should be noted that compensatory mechanisms aimed at restoration of NO bioavailability in conditions of severely running infectious process, as a rule, are untenable. Thus, in case of a pronounced inflammatory reaction, endothelial cells and macrophages/monocytes begin to synthesize inducible nitric oxide synthase (iNOS) [48]. However, this isoform of the enzyme generates much higher concentrations of NO, which causes the predominance of indirect effects associated with the formation of peroxynitrite and free-radical damage of cells [49].
Cytokinemia and the cytokine storm
Using highly pathogenic influenza A(H5N1) virus as an example, it was shown that during infection of lung microvascular endothelial cells, both a more pronounced activation of the transcription factor NF-κB and additional activation of mitogen-activated protein kinase (MAPK) signaling pathways are observed [50]. In particular, highly pathogenic influenza viruses activate the p38 MAPK pathway, which is manifested by excessive and uncontrolled synthesis of pro-inflammatory cytokines. It is important to note that in this case it is the activated endothelium of pulmonary microvessels that becomes the main producer of pro-inflammatory cytokines (interleukins (IL) 1β, IL-6, tumor necrosis factor-alpha (TNF-α)) and chemokines (CXCL10, RANTES) [42, 51, 52]. In addition, the resulting hypercytokinemia mediates systemic damage to the endothelium of other vascular regions, which is the cause of the development of a cytokine storm and ARDS.
It should be emphasized that the KLF2 transcription factor plays an important role in the development of the cytokine storm in influenza. Under physiological conditions, this transcription factor maintains NO bioavailability at an optimal level, as well as barrier properties and thromboresistance of endothelial cells [53‒55]. The results of the study by R. Huang et al. showed that during infection with highly pathogenic influenza viruses in BALB/c mice the level of expression of the transcription factor KLF2 in the endothelium of lung microvessels is significantly reduced, which accounts for the development of cytokine storm, acute lung injury and ARDS. These changes correlate with the fact that with excessive activation of the transcription factor NF-κB, there is a decrease in the activity of the KLF2 factor [56].
Disruption of metabolic processes
IAVs, including subtypes A(H1N1)pdm09, A(H3N2) and A(H5N1), depress metabolic processes in endothelial cells and cause a twofold decrease in dehydrogenase activity. It should be noted that a decrease in the activity of dehydrogenases, but to a lesser extent, is also observed in response to the introduction of separate surface proteins of different IAV subtypes, hemagglutinin and neuraminidase, into the culture medium [57].
It is likely that one of the reasons for such pronounced changes in metabolic processes is the effect on endothelial cells of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, which can suppress mitochondrial activity and, consequently, catabolic processes [58]. In addition, the formation of intracellular peroxynitrite against the background of dysregulation of eNOS expression causes damage to mitochondrial membranes, which also contributes to the decrease in mitochondrial activity.
Disruption of barrier properties, changes in morphology and increased endothelial permeability
Intact glycocalyx plays an important role in the maintenance of endothelial barrier properties. The glycocalyx of vascular endothelium mainly consists of proteoglycans and glycoproteins bound to sialic acids [59]. The enzymatic activity of influenza virus neuraminidase allows to cleave the bond between the terminal residue of sialic acid and glycoprotein in the glycocalyx of endotheliocytes, which causes its thinning and detachment. Furthermore, IAV-activated endothelium begins to synthesize matrix metalloproteinases-2 and -9, which increase damage of the glycocalyx. As a result, the endothelial glycocalyx undergoes degradation, loses its negative charge, as a result of which the endothelium becomes permeable to molecules with relatively high molecular weight [60].
Endothelial permeability also depends on the integrity of intercellular contacts (tight, adhesive, and gap junctions) [61]. In vitro studies have shown that IAVs cause degradation of various proteins that are part of intercellular contacts (β-catenin, claudin-5, VE-cadherin, ZO-1 protein, and connexins), as well as reorganization of the actin cytoskeleton of endotheliocytes with the formation of stress-induced fibrils [58, 62]. As a consequence, endothelial cells shrink and round with the formation of gaps between them, which is the cause of increased vascular permeability [42].
It should be noted that damage of endothelial glycocalyx, as well as degradation of proteins, which are part of intercellular contacts, can be observed when endothelial cells are exposed to high concentrations of cytokines (IL-1β and TNF-α), which also significantly increases endothelial permeability of lung microvessels and underlies the development of acute lung injury and ARDS [63‒66].
Disruption of adhesive properties
Activation of the NF-κB signaling pathway in influenza leads to increased expression of various cell adhesion molecules, including adhesive receptors of the immunoglobulin superfamily (ICAM-1, ICAM-2, VCAM-1, PECAM-1) and selectins (P-selectin, E-selectin) [20, 67‒69].
In turn, increased adhesive properties allow circulating leukocytes to perform adhesion on endothelial cells and then migrate from the vascular channel to the focus of inflammation. However, in the systemic inflammatory reaction against the background of severe influenza, there is a pronounced and unregulated adhesion of leukocytes to the vascular endothelium, and the severity of the course of influenza depends largely on the degree of involvement of neutrophils in the inflammatory reaction [70]. Neutrophils in severe influenza become one of the main producers of ROS and inflammatory mediators, and also synthesize neutrophil extracellular traps (NETs), mediating cell damage [71‒73].
Disruption of hemostasis and fibrinolysis
The severe course of influenza is characterized by disorders of coagulation and fibrinolysis by the type of DIC [29]. IAVs mediate activation of both external and internal pathways of coagulation (Fig. 2). In response to infection or as a result of exposure of endothelial cells to high concentrations of pro-inflammatory cytokines (TNF and IL-1β), the expression of endothelial factors with pro-coagulatory properties increases in cells: vWF, platelet activation factor (PAF), plasminogen activator inhibitor-1 (PAI-1), and tissue factor (TF), a key glycoprotein that triggers the extrinsic coagulation pathway [20, 74‒77]. The studies by F. Visseren et al. showed that infection of endothelial cells with influenza A(H1N1) and A(H3N2) viruses increases TF expression, which reduces blood clotting time by 55% within 3 h after infection [78]. Similar data on the increase in TF concentration in bronchoalveolar lavage were obtained when BALB/c mice were infected with the A(H1N1) virus [79].

Fig. 2. The coagulation cascades.
1 ‒ intact endothelial cells express antiplatelet and anticoagulant agents (thrombomodulin, antithrombin, tissue factor pathway inhibitor and ADPase) that prevent aggregation of platelet and fibrin formation; 2 ‒ coagulation is usually initiated by an injury to the endothelium, with the exposure of tissue factor and collagen from the subendothelium to the blood factors and the release of von Willebrand factor (vWF); 3 ‒ activation of platelets is initiated by exposure to tissue factor, collagen and vWF. Activated platelets release several mediators (including ADP and vWF), leading to further platelet recruitment, activation, aggregation and plug formation (primary hemostasis); 4 ‒ the extrinsic pathway is initiated by the interaction between tissue factor and Factor VII; 5 ‒ the intrinsic pathway is initiated by the exposure of collagen to Factor XII; 6 ‒ the extrinsic and intrinsic coagulation pathways lead into the final common pathway, which contains cascades involved in the production of thrombin, activated Factor X and the formation of fibrin strands; 7 ‒ fibrin strands increase stability of the platelet plug and lead to the formation of platelet-fibrin clot (secondary hemostasis); 8 ‒ kallikrein, tissue plasminogen activator (tPA) or urokinase plasminogen activator (uPA) convert plasminogen to plasmin, which then degrades and reabsorbs the fibrin strands in process called fibrinolysis. Endothelial factors whose concentration in influenza A virus infection is reliably changed are underlined (see Table).
Рис. 2. Каскад коагуляции.
1 ‒ интактные эндотелиальные клетки (ЭК) обеспечивают тромборезистентность за счет постоянного синтеза ряда антикоагулянтов (тромбомодулина, антитромбина, ингибитора пути тканевого активатора и АДФазы); 2 ‒ каскад коагуляции, как правило, возникает вследствие повреждения ЭК и связан с активацией факторов свертывания крови при контакте с тканевым фактором и коллагеном из субэндотелиальной слоя, а также с высвобождением из клеток фактора Виллебранда (vWF); 3 ‒ активация тромбоцитов возникает при взаимодействии с тканевым фактором, коллагеном и vWF. Активированные тромбоциты высвобождают ряд медиаторов, таких как АДФ и vWF, что приводит к дальнейшему рекрутингу, активации и агрегации тромбоцитов с образованием первичного тромбоцитарного сгустка (первичный гемостаз); 4 ‒ внешний путь свертывания инициируется при контакте фактора VII с тканевым фактором; 5 ‒ внутренний путь инициируется при контакте фактора XII с коллагеном; 6 ‒ внешний и внутренний пути приводят к инициированию общего пути свертывания, который содержит каскады, участвующие в активации фактора X и тромбина с образованием нитей фибрина; 7 ‒ нити фибрина способствуют повышению стабильности тромбоцитарного сгустка и приводят к образованию тромбоцитарно-фибринового сгустка (вторичный гемостаз); 8 ‒ калликреин, тканевой активатор плазминогена (tPA) или урокиназный активатор плазминогена (uPA) конвертируют плазминоген в плазмин, который затем разрушает и реабсорбирует полимеризованные нити фибрина, что необходимо для разрушения сгустков в рамках процесса фибринолиза. Подчеркнуты эндотелиальные факторы, концентрация в крови которых при гриппозной инфекции достоверно изменяется (см. таблицу).
The important role of factor PAI-1 in the pathogenesis of influenza should be noted. This endothelial factor is an antagonist of two proteins of the fibrinolysis system – urokinase and plasminogen TF (uPA and tPA), which carry out the cleavage of plasminogen to plasmin [80]. In turn, plasmin can be used by influenza viruses to hydrolyze hemagglutinin precursor protein (HA0), which is necessary for maturation of newly synthesized virions. Thus, increased expression of PAI-1, on the one hand, reduces the infectious activity of the virus, and on the other hand, may be the cause of inhibition of fibrinolysis and increased thrombosis in influenza.
In vivo activation of the extrinsic and intrinsic pathway occurs when endothelial cells are damaged. In particular, TF and collagen located subendothelially as a result of endothelial desquamation begin to interact with factors VII and XII, which is necessary for activation of the common pathway of coagulation with platelet activation and formation of platelet clots. [29]. Thus, the degree of vascular endothelial damage probably correlates with the severity of activation of the coagulation system in influenza.
In addition to taking part in hemostasis, platelets also play an important role in the immune response. For example, platelets are able to capture and sequester viruses, which helps to limit their spread [81]. However, it is likely that endocytosis of influenza viruses is not always complete. In their studies, M. Koupenova et al. found that some fragments of viral particles remain exposed on the surface of platelets. This, in turn, may mediate a longer contact of vascular endothelial cells with pathogen-associated molecular patterns of the virus, leading to a more pronounced activation of endotheliocytes.
Depletion of pro-coagulative factors and the developing thrombocytopenia may subsequently lead to a hypocoagulation phase [82]. Influenza viruses due to neuraminidase activity can mediate the cleavage of sialic acids from the glycocalyx of platelets, which enhances their removal from the bloodstream up to the development of thrombocytopenia [83]. Furthermore, the severity of thrombocytopenia depends on the virus subtype. Thus, influenza A(H5N1) virus causes more pronounced thrombocytopenia compared to IAV subtypes A(H1N1) and A(H3N2).
Disruption of immune processes
The infected vascular endothelium is unable to adequately support immune processes, primarily related to antigen presentation to immunocompetent cells. In addition, there is also a dysregulation in the activation of cells of innate immunity [84]. As noted previously, IL-1β synthesis is significantly increased in IAV-infected endothelial cells [85]. This pro-inflammatory cytokine plays an important role in the innate immune response, as it mediates the process of antigen-dependent differentiation of T-lymphocytes, as well as the differentiation of dendritic cells. Thus, in severe influenza with the involvement of vascular endothelial cells in the inflammatory process due to the synthesis of IL-1β there is an excessive infiltration of pulmonary parenchyma by immune cells, which may cause acute lung injury.
Disruption of angiogenesis processes
Changes in the expression level of endothelial factors affecting angiogenesis are not characteristic of influenza infection [86]. However, the results of the study by S. Morichi et al. showed that in children during the development of virus-induced encephalopathy in cerebrospinal fluid there is an increase in the expression of two markers of angiogenesis – vascular endothelial growth factor (VEGF-A) and platelet-derived growth factor (PDGF).
Apoptosis
When endotheliocytes are infected with IAV, cell apoptosis can occur due to activation of both receptor-dependent (external) and mitochondrial (internal) signaling pathways. One of the reasons for activation of the extrinsic pathway of apoptosis is exposure of the cell to high concentrations of TNF-α. This is confirmed by the fact that the expression of caspase-3, an effector protease that cleaves cytoskeleton and activates endonuclease at the terminal stages of apoptosis, increases in infected endothelial cells [26, 87].
The intrinsic pathway of apoptosis activation is realized by mitochondrial membrane damage. Intracellular reactive oxygen and nitrogen species, in particular peroxynitrite, mediate damage to the membranes of these organelles with the release of cytochrome c [88]. Furthermore, the main protein of influenza virus M1 is able to bind to an important component of cytoprotection – the heat shock protein 70 (Hsp70), resulting in dissociation of Hsp70 and APAF-1 protein with the release of the latter and formation of apoptosomes [89]. As a result, virus-induced apoptosis leads to the disruption of the integrity of the endothelial barrier, which causes increased vascular permeability and plays a key role in the pathogenesis of thrombosis, DIC, vasculitis and atherosclerosis [62, 90, 91].
It should be noted that in viral infections, early activation of apoptosis in most cases makes it possible to significantly suppress viral replication, whereas delayed apoptosis of infected cells causes the spread of viral particles in the body [92]. In turn, with respect to influenza virus, in vitro studies have shown that apoptosis of infected cells is delayed due to the anti-apoptotic activity of the viral protein NS1 [93, 94]. Furthermore, in addition to apoptosis, IAV is capable of inducing other programmed cell death variants, including: 1) necroptosis (due to HA, NS1, PB1 proteins); 2) pyroptosis (due to M2, PB1-F2, PB2 proteins); 3) autophagy (due to HA protein) [92, 95].
Based on the above, it becomes evident that in the severe course of influenza infection caused by IAV, there is a violation of all endothelial functions: vasomotor, adhesive, hemostatic, angiogenic, immune and others (Table). As the main causes of ED development, it is likely to be considered the impact on the endothelium of several phlogogenic factors at once, among which the cytopathic effect of the virus, oxidative stress, cytokinemia and hypoxia are of the greatest importance. Furthermore, not only the virus itself, but also its proteins (e.g., hemagglutinin and neuraminidase) can cause marked activation of endotheliocytes, which may cause the development of their dysfunction [57].
Table. Alteration of vascular endothelium functions caused by Influenza A Viruses
Таблица. Нарушения функций сосудистого эндотелия, вызванные вирусами гриппа типа А
Functions of endothelial cells Функции клеток эндотелия | Endothelial factors Эндотелиальные факторы | Reference Источник |
Vasomotor Вазомоторная | NO (by eNOS) ↓ NO (за счет eNOS) ↓ | |
Adhesion Адгезивная | ICAM-1 ↑ | |
P-selectin/P-селектин ↑ | ||
E-selectin/E-селектин ↑ | [98] | |
PECAM-1 ↑ | ||
Hemostatic Гемостатическая | tPA ↔ / ↑ | |
PAI-1 ↔ / ↑ | ||
vWF ↑ | [20] | |
PAF ↑ | [99] | |
TF ↑ | ||
Angiogenic Ангиогенная | VEFG ↑ PDGF ↑ | [100] |
Immune Иммунная | IL-1 ↑ |
Note. ↔ – modulation of the expression; ↑ – increased expression; ↓ – decreased expression. ICAM-1 – Intercellular adhesion molecule-1; PECAM-1 – platelet and endothelial cell adhesion molecule-1; tPA – tissue plasminogen activator; PAI-1 – plasminogen activator inhibitor-1; vWF – von Willebrand factor; PAF – platelet-activating factor; TF – tissue factor; VEFG – vascular endothelial growth factor; PDGF – platelet-derived growth factor; IL-1 – Interleukin-1.
Примечание. ↔ – модуляция экспрессии; ↑ – повышение экспрессии; ↓ – снижение экспрессии. ICAM-1 – межклеточная молекула адгезии-1; PECAM-1 – молекула адгезии тромбоцитов и эндотелиоцитов-1; tPA – тканевой активатор плазминогена; PAI-1 – ингибитор активатора плазминогена-1; vWF – фактор Виллебранда; PAF – фактор активации тромбоцитов; TF – тканевой фактор; VEFG – фактор роста эндотелия сосудов; PDGF – фактор роста тромбоцитов; IL-1 – интерлейкин-1.
Molecular mimicry
It can be assumed that molecular mimicry is also a probable cause of ED in influenza. Thus, in different strains of influenza viruses A(H1N1)pdm09, as well as A(H3N2) (data not published), many amino acid sequences have been found in proteins with a high degree of homology with amino acid sequences in various proteins of the hemostasis system and endothelial factors, including vWF, eNOS, PAI-1, TF, tPA, blood coagulation factors (III, V, VI, VII, VIII, VIII, IX, X, XI, XIII) and others. In particular, numerous sequences of 12 amino acid residues in length with homology exceeding 80% were found in the above proteins and various proteins of influenza viruses [103].
Bioinformatics analysis allowed us to detect a number of unique sequences in the composition of influenza A/H1N1 virus, which caused the 1918‒1920 pandemic, mimicking fragments in the composition of proteins of the hemostasis and fibrinolysis system (fibrinogen, tissue factor, antithrombin-III, prothrombin, plasminogen, urokinase plasminogen activator, etc.), which are absent in the influenza A(H1N1)pdm09 virus isolated in 2016 [92].
Interestingly, when comparing modern circulating strains of influenza A(H1N1)pdm09 and A(H3N2) viruses, the latter have a large number of amino acid sequences homologous to the sequences of various factors of the fibrinolysis system (α2-antiplasmin, α2-macroglobulin, thrombomodulin, urokinase plasminogen activator and kallikrein). It is worth noting that the conservation of these sequences, both in terms of location and amino acid composition, has been preserved in the proteins of A(H3N2) viruses for 50 years. This feature probably allows influenza A(H3N2) viruses to cause a more intense epidemic process (compared to A(H1N1)pdm09) and to cause a higher level of additional mortality from influenza in patients with concomitant cardiovascular diseases [104]. Thus, the presence of mimicry sequences in proteins of the hemostasis system and influenza viruses, apparently, increases their virulence, because it allows to disrupt the processes of coagulation and fibrinolysis.
It is known that the severity of ED in influenza depends not only on the virus (virulence of the strain, infectious dose) and resistance of the organism (pre-existing immunity, genetic predisposition to the development of severe forms of the disease), but also on the state of the cardiovascular system. Thus, the presence of acute and chronic diseases of the cardiovascular system in patients causes a more severe course of influenza, which is associated with the involvement of the vascular endothelium in the pathological process. This is confirmed by the fact that after the end of the epidemic in patients with cardiovascular diseases the rate of additional mortality from influenza is 481 per 100 thousand of the population against 2 per 100 thousand of the population among healthy adults without somatic diseases.
Conclusion
IAVs can cause activation and dysfunction of the vascular endothelium, which is a key link in the pathogenesis of severe influenza, causing the development of complications in the acute and delayed period of infection. In turn, ED is both a risk factor and a key link in the pathogenesis of many diseases of the cardiovascular system, including atherosclerosis, arterial hypertension, myocardial infarction, ischemic stroke and other pathologies.
Despite the availability of vaccination as the main measure of influenza prevention and a number of etiotropic drugs from different clinical-pharmacological groups that effectively inhibit various stages of virus reproduction, an important direction remains the optimization of pathogenetic therapy of influenza. Taking into account the high risk of severe influenza in patients from risk groups, especially in patients over 65 years old with chronic diseases of cardiovascular system, it is reasonable to prescribe chemical medicines with proven endothelioprotective activity to correct endothelial dysfunction as part of pathogenetic therapy.
About the authors
Vladimir A. Marchenko
North-Western State Medical University Named after I.I. Mechnikov
Author for correspondence.
Email: vmarcenco@mail.ru
ORCID iD: 0000-0001-6870-3157
Ph. D. in medicine, Associate Professor of Medical Microbiology Department
Россия, 191015, St. PetersburgIrina N. Zhilinskaya
North-Western State Medical University Named after I.I. Mechnikov
Email: vmarcenco@mail.ru
ORCID iD: 0000-0002-0084-1323
D. Sc. in Biology, Professor of Medical Microbiology Department
Россия, 191015, St. PetersburgReferences
- Office WHOEMR. Global Influenza Strategy 2019–2030. Weekly Epidemiological Record; 2019.
- Boehme A.K., Luna J., Kulick E.R., Kamel H., Elkind M.S.V. Influenza-like illness as a trigger for ischemic stroke. Ann. Clin. Transl. Neurol. 2018; 5(4): 45663. https://doi.org/10.1002/acn3.545
- Muscente F., De Caterina R. Causal relationship between influenza infection and risk of acute myocardial infarction: pathophysiological hypothesis and clinical implications. Eur. Heart J. 2020; 22(Suppl. E): E68–72. https://doi.org/10.1093/eurheartj/suaa064
- Skaarup K.G., Modin D., Nielsen L., Jensen J.U.S., Biering-Sørensen T. Influenza and cardiovascular disease pathophysiology: strings attached. Eur. Heart J. 2023;25(Suppl. A): A5–11. https://doi.org/10.1093/eurheartjsupp/suac117
- Rubino R., Imburgia C., Bonura S., Trizzino M., Iaria C., Cascio A. Thromboembolic events in patients with influenza: a scoping review. Viruses. 2022; 14(12): 2817. https://doi.org/10.3390/v14122817
- Short K.R., Kuiken T., Van Riel D. Role of endothelial cells in the pathogenesis of influenza in humans. J. Infect. Dis. 2019; 220(11): 1859–60. https://doi.org/10.1093/infdis/jiz349
- Armstrong S.M., Darwish I., Lee W.L. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence. 2013; 4(6): 537–42. https://doi.org/10.4161/viru.25779
- Marchenko V.A., Barashkova S.V., Zelinskaya I.A., Toropova Ya.G., Ramsay E.S., Zhilinskaya I.N. Modulation of endothelial factors activity in human endothelial cells in influenza A(H1N1)PDM09 virus infection. Voprosy virusologii. 2021; 66(3): 198–210. https://doi.org/10.36233/0507-4088-48 https://elibrary.ru/wsxlvb (in Russian)
- Aird W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007; 100(2): 158–73. https://doi.org/10.1161/01.RES.0000255691.76142.4a
- Zhang J., Defelice A.F., Hanig J.P., Colatsky T. Biomarkers of endothelial cell activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol. Pathol. 2010; 38(6): 856–71. https://doi.org/10.1177/0192623310378866
- Matrosovich M.N., Matrosovich T.Y., Gray T., Roberts N.A., Klenk H.D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA. 2004; 101(13): 4620–4. https://doi.org/10.1073/pnas.0308001101
- Ibricevic A., Pekosz A., Walter M.J., Newby C., Battaile J.T., Brown E.G., et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006; 80(15): 7469–80. https://doi.org/10.1128/JVI.02677-05
- Abe Y., Smith C.W., Katkin J.P., Thurmon L.M., Xu X., Mendoza L.H., et al. Endothelial alpha 2,6-linked sialic acid inhibits VCAM-1-dependent adhesion under flow conditions. J. Immunol. 1999; 163(5): 2867–76.
- Cioffi D.L., Pandey S., Alvare D.F., Cioffi E.A. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell Mol. Physiol. 2012; 302(10): L1067–77. https://doi.org/10.1152/ajplung.00190.2011
- Denney L., Ho L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018; 41(4): 218–33. https://doi.org/10.1016/j.bj.2018.08.004
- Herold S., Becker C., Ridge K.M., Budinger G.R. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur. Respir. J. 2015; 45(5): 1463–78. https://doi.org/10.1183/09031936.00186214
- Herold S., Steinmueller M., von Wulffen W., Cakarova L., Pinto R., Pleschka S., et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008; 205(13): 3065–77. https://doi.org/10.1084/jem.20080201
- Zeng H., Goldsmith C.S., Maines T.R., Belser J.A., Gustin K.M., Pekosz A., et al. Tropism and infectivity of influenza virus, including highly pathogenic avian H5N1 virus, in ferret tracheal differentiated primary epithelial cell cultures. J. Virol. 2013; 87(5): 2597–607. https://doi.org/10.1128/JVI.02885-12
- Kumlin U., Olofsson S., Dimock K., Arnberg N. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir. Viruses. 2008; 2(5): 147–54. https://doi.org/10.1111/j.1750-2659.2008.00051.x
- Sugiyama M.G., Gamage A., Zyla R., Armstrong S.M., Advani S., Advani A., et al. Influenza virus infection induces platelet-endothelial adhesion which contributes to lung injury. J. Virol. 2015; 90(4): 1812–23. https://doi.org/10.1128/JVI.02599-15
- Lee S., Hirohama M., Noguchi M., Nagata K., Kawaguchi A. Influenza A virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the type I interferon signaling pathway in a mutually exclusive manner. J. Virol. 2018; 92(14): e00396-18. https://doi.org/10.1128/JVI.00396-18
- Chan M.C., Chan R.W., Yu W.C., Ho C.C., Chui W.H., Lo C.K., et al. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir. Res. 2009; 10(1): 102. https://doi.org/10.1186/1465-9921-10-102
- Zeng H., Pappas C., Belser J.A., Houser K.V., Zhong W., Wadford D.A., et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J. Virol. 2012; 86(2): 667–78. https://doi.org/10.1128/JVI.06348-11
- Chan L.L.Y., Hui K.P.Y., Kuok D.I.T., Bui C.H.T., Ng K.C., Mok C.K.P., et al. Risk assessment of the tropism and pathogenesis of the highly pathogenic avian influenza A/H7N9 virus using ex vivo and in vitro cultures of human respiratory tract. J. Infect. Dis. 2019; 220(4): 578–88. https://doi.org/10.1093/infdis/jiz165
- Simon P., de La Vega M.A., Paradis É., Mendoza E., Coombs K.M., Kobasa D., et al. Avian influenza viruses that cause highly virulent infections in humans exhibit distinct replicative properties in contrast to human H1N1 viruses. Sci. Rep. 2016; 6: 24154. https://doi.org/10.1038/srep24154
- Han T., Lai Y., Jiang Y., Liu X., Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading to microvascular leakage and release of pro-inflammatory cytokines. PeerJ. 2021; 9: e11892. https://doi.org/10.7717/peerj.11892
- Gu Y., Zuo X., Zhang S., Ouyang Z., Jiang S., Wang F., et al. The mechanism behind influenza virus cytokine storm. Viruses. 2021; 13(7): 1362. https://doi.org/10.3390/v13071362
- Tang B.M., Cootes T., McLean A.S. From influenza-induced acute lung injury to multiorgan failure. In: Annual Update in Intensive Care and Emergency Medicine 2019. 2018: 449–58. https://doi.org/10.1007/978-3-030-06067-1_35
- Yang Y., Tang H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016; 13(4): 432–42. https://doi.org/10.1038/cmi.2016.1
- Zhang J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc. Med. 2022; 23(2): 73. https://doi.org/10.31083/j.rcm2302073
- Immanuel J., Yun S. Vascular inflammatory diseases and endothelial phenotypes. Cells. 2023; 12(12): 1640. https://doi.org/10.3390/cells12121640
- Mel’nikova Yu.S., Makarova T.P. Endothelial dysfunction as the key link of chronic diseases pathogenesis. Kazanskii meditsinskii zhurnal. 2015; 96(4): 659–65. https://doi.org/10.17750/KMJ2015-659 https://elibrary.ru/ubegwv (in Russian)
- Vlasova T.I., Petrishchev N.N., Vlasov T.D. Endothelial dysfunction as the typical pathological state. Regionarnoe krovoobrashchenie i mikrotsirkulyatsiya. 2022; 21(2): 4–15. https://doi.org/10.24884/1682-6655-2022-21-2-4-15 https://elibrary.ru/zheshs (in Russian)
- Yang Y., Bazhin A.V., Werner J., Karakhanova S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013; 32(3): 249–70. https://doi.org/10.3109/08830185.2012.755176
- Bach F.H., Robson S.C., Ferran C., Winkler H., Millan M.T., Stuhlmeier K.M., et al. Endothelial cell activation and thromboregulation during xenograft rejection. Immunol. Rev. 1994; 141: 5–30. https://doi.org/10.1111/j.1600-065x.1994.tb00870.x
- Pober J.S., Sessa W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007; 7(10): 803–15. https://doi.org/10.1038/nri2171
- Bigildeev A.E., Chepurnykh Yu.F., Petinati N.A., Drize N.J. Features of the expression of NF-kB pathway genes in tissues of irradiated mice and in old animals. Radiatsionnaya biologiya. Radioekologiya. 2019; 59(6): 565–74. https://doi.org/10.1134/S0869803119060031 https://elibrary.ru/ebdunp (in Russian)
- Waitkus M.S., Harris D.P., DiCorleto P.E. Mechanisms of Endothelial Activation. In: Mackay I.R., Rose N.R., Diamond B., Davidson A., eds. Encyclopedia of Medical Immunology. New York: Springer; 2014. https://doi.org/10.1007/978-0-387-84828-0_183
- Endemann D.H., Schiffrin E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004; 15(8): 1983–92. https://doi.org/10.1097/01.ASN.0000132474.50966.DA
- Hadi H.A., Carr C.S., Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005; 1(3): 183–98.
- Widmer R.J., Lerman A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014; 2014(3): 291–308. https://doi.org/10.5339/gcsp.2014.43
- Han T., Lai Y., Jiang Y., Liu X., Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading to microvascular leakage and release of pro-inflammatory cytokines. PeerJ. 2021; 9: e11892. https://doi.org/10.7717/peerj.11892
- Siragusa M., Thole J., Bibli S.I., Luck B., Loot A.E., de Silva K., et al. Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J. 2019; 38(17): e100938. https://doi.org/10.15252/embj.2018100938
- Marchenko V.A., Zelinskaya I.A., Toropova YA.G., Mukhametdinova D.V., Galagudza M.M., Lioznov D.A., et al. Duration of systemic alteration in vasomotor function of microvascular endothelium caused by the influenza A(H1N1)pdm09 virus. Regionarnoe krovoobrashchenie i mikrotsirkulyatsiya. 2023; 22(4): 74–86. https://doi.org/10.24884/1682-6655-2023-22-4-74-86 https://elibrary.ru/mmwnsf (in Russian)
- Boytsov SA. Influenza, novel coronavirus infection and cardiovascular diseases. Russian Kardiologicheskii vestnik. 2021; 16(1): 5–9. https://doi.org/10.17116/Cardiobulletin2021160115 https://elibrary.ru/zgvxkg (in Russian)
- Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA. 2018; 115(23): 5839–48. https://doi.org/10.1073/pnas.1804932115
- Beckman J.S., Koppenol W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996; 271(5 Pt. 1): C1424–37. https://doi.org/10.1152/ajpcell.1996.271.5.C1424.
- Babizhayev M.A., Deyev A.I. Management of the virulent influenza virus infection by oral formulation of nonhydrolized carnosine and isopeptide of carnosine attenuating proinflammatory cytokine-induced nitric oxide production. Am. J. Ther. 2012; 19(1): e25–47. https://doi.org/10.1097/MJT.0b013e3181dcf589
- Vasina L.V., Petrishchev N.N., Vlasov T.D. Markers of endothelial dysfunction. Regionarnoe krovoobrashchenie i mikrotsirkulyatsiya. 2017; 16(1): 4–15. https://doi.org/10.24884/1682-6655-2017-16-1-4-15 https://elibrary.ru/yocujf (in Russian)
- Viemann D., Schmolke M., Lueken A., Boergeling Y., Friesenhagen J., Wittkowski H., et al. H5N1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J. Immunol. 2011; 186(1): 164–73. https://doi.org/10.4049/jimmunol.0904170
- Teijaro J.R., Walsh K.B., Cahalan S., Fremgen D.M., Roberts E., Scott F., et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011; 146(6): 980–91. https://doi.org/10.1016/j.cell.2011.08.015
- Yu J., Sun X., Goie J.Y.G., Zhang Y. Regulation of host immune responses against influenza A virus infection by Mitogen-Activated Protein Kinases (MAPKs). Microorganisms. 2020; 8(7): 1067. https://doi.org/10.3390/microorganisms8071067
- Fontijn R.D., Volger O.L., van der Pouw-Kraan T.C., Doddaballapur A., Leyen T., Baggen J.M., et al. Expression of nitric oxide-transporting aquaporin-1 is controlled by KLF2 and marks non-activated endothelium in vivo. PLoS One. 2015; 10(12): e0145777. https://doi.org/10.1371/journal.pone.0145777
- Parmar K.M., Larman H.B., Dai G., Zhang Y., Wang E.T., Moorthy S.N., et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 2006; 116(1): 49–58. https://doi.org/10.1172/jci24787
- SenBanerjee S., Lin Z., Atkins G.B., Greif D.M., Rao R.M., Kumar A., et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004; 199: 1305–15. https://doi.org/10.1084/jem.20031132
- Turpaev K.T. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochemistry (Mosc.). 2020; 85(1): 54–67. https://doi.org/10.1134/S0006297920010058
- Azarenok A.A., Eropkina E.M., Prochukhanova A.R., Shaldzhyan A.A., Kozlova N.M., Kozeletskaya K.N., et al. The influenza viruses and their surface proteins impact on the metabolism of human blood vessel endothelium cells. Voprosy virusologii. 2013; 58(3): 25–7. https://elibrary.ru/pzxtur (in Russian)
- Hiyoshi M., Indalao I.L., Yano M., Yamane K., Takahashi E., Kido H. Influenza A virus infection of vascular endothelial cells induces GSK-3β-mediated β-catenin degradation in adherens junctions, with a resultant increase in membrane permeability. Arch. Virol. 2015; 160(1): 225–34. https://doi.org/10.1007/s00705-014-2270-5
- Betteridge K.B., Arkill K.P., Neal C.R., Harper S.J., Foster R.R., Satchell S.C., et al. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. J. Physiol. 2017; 595(15): 5015–35. https://doi.org/10.1113/JP274167
- Taghavi S., Abdullah S., Shaheen F., Mueller L., Gagen B., Duchesne J., et al. Glycocalyx degradation and the endotheliopathy of viral infection. PLoS One. 2022; 17(10): e0276232. https://doi.org/10.1371/journal.pone.0276232
- Simionescu M. Structural biochemical and functional differentiation of the vascular endothelium. In: Risau W., ed. Morphogenesis of the Endothelium. Amsterdam: Harwood Academic; 2000: 1–21.
- Armstrong S.M., Wang C., Tigdi J., Si X., Dumpit C., Charles S., et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS One. 2012; 7(10): e47323 https://doi.org/10.1371/journal.pone.0047323
- Yang Y., Schmidt E.P. The endothelial glycocalyx: an important regulator of the pulmonary vascular barrier. Tissue Barriers. 2013; 1(1): e23494. https://doi.org/10.4161/tisb.23494
- Ferro T., Neumann P., Gertzberg N., Clements R., Johnson A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000; 278(6): L1107–17. https://doi.org/10.1152/ajplung.2000.278.6.L1107.
- Kim K., Jung H., Shin I., Choi B., Kim D. Induction of interleukin-1 beta (IL-1 β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015; 87: 1104–12. https://doi.org/10.1002/jmv.24138.
- Wang S., Le T.Q., Kurihara N., Chida J., Cisse Y., Yano M., et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J. Infect. Dis. 2010; 202(7): 991–1001. https://doi.org/10.1086/656044
- Collins T., Read M.A., Neish A.S., Whitley M.Z., Thanos D., Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995; 9(10): 899-909.
- Guan X., Yang W., Sun X., Wang L., Ma B., Li H., et al. Association of influenza virus infection and inflammatory cytokines with acute myocardial infarction. Inflamm. Res. 2012; 61(6): 591–8. https://doi.org/10.1007/s00011-012-0449-3
- Singh V., Kaur R., Kumari P., Pasricha C., Singh R. ICAM-1 and VCAM-1: Gatekeepers in various inflammatory and cardiovascular disorders. Clin. Chim. Acta. 2023; 548: 117487. https://doi.org/10.1016/j.cca.2023.117487
- George S.T., Lai J., Ma J., Stacey H.D., Miller M.S., Mullarkey C.E. Neutrophils and influenza: a thin line between helpful and harmful. Vaccines (Basel). 2021; 9(6): 597. https://doi.org/10.3390/vaccines9060597
- Tang B.M., Shojaei M., Teoh S., Meyers A., Ho J., Ball T.B., et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat. Commun. 2019; 10(1): 3422. https://doi.org/10.1038/s41467-019-11249-y
- Narasaraju T., Yang E., Samy R.P., Ng H.H., Poh W.P., Liew A.A., et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011; 179(1): 199–210. https://doi.org/10.1016/j.ajpath.2011.03.013
- Saffarzadeh M., Juenemann C., Queisser M.A., Lochnit G., Barreto G., Galuska S.P., et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012; 7(2): e32366; http://doi.org/10.1371/journal.pone.0032366
- Schleef R.R., Bevilacqua M.P., Sawdey M., Gimbrone M.A. Jr., Loskutoff D.J. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J. Biol. Chem. 1988; 263(12): 5797–803.
- Marchenko V., Mukhametdinova D., Amosova I., Lioznov D., Zhilinskaya I. Influenza A(H1N1)pdm09 virus alters expression of endothelial factors in pulmonary vascular endothelium in rats. Viruses. 2022; 14(11): 2518. https://doi.org/10.3390/v14112518
- Bussolino F., Camussi G., Baglioni C. Synthesis and release of platelet-activating factor by human vascular endothelial cells treated with tumor necrosis factor or interleukin 1 alpha. J. Biol. Chem. 1988; 263(24): 11856–61.
- Schastlivtsev I.V., Lobastov K.V., Tsaplin S.N., Mkrtychev D.S. Modern view on hemostasis system: cell theory. Meditsinskii sovet. 2019; (16): 72–7. https://doi.org/10.21518/2079-701X-2019-16-72-77 https://elibrary.ru/smgyfk (in Russian)
- Visseren F.L., Bouwman J.J., Bouter K.P., Diepersloot R.J., de Groot P.H., Erkelens D.W. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb. Haemost. 2000; 84(2): 319–24.
- Zelaya H., Tada A., Vizoso-Pinto M.G., Salva S., Kanmani P., Agüero G., et al. Nasal priming with immunobiotic Lactobacillus rhamnosus modulates inflammation-coagulation interactions and reduces influenza virus-associated pulmonary damage. Inflamm. Res. 2015; 64(8): 589–602. https://doi.org/10.1007/s00011-015-0837-6
- Cesari M., Pahor M., Incalzi R.A. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010; 28(5): e72–91. https://doi.org/10.1111/j.1755-5922.2010.00171.x
- Slukhanchuk E.V., Bitsadze V.O., Khizroeva J.KH., Solopova A.G., Tsibizova V.I., Yakubova F., et al. The role of platelets in antiviral immunity. Akusherstvo, ginekologiya i reproduktsiya. 2022; 16(2): 204–12. https://doi.org/10.17749/2313-7347/ob.gyn.rep.2022.305 https://elibrary.ru/twhjna (in Russian)
- Iba T., Levi M., Thachil J., Levy J.H. Disseminated intravascular coagulation: the past, present, and future considerations. Semin. Thromb. Hemost. 2022; 48(8): 978–87. https://doi.org/10.1055/s-0042-1756300
- Jansen A.J.G., Spaan T., Low H.Z., Di Iorio D., van den Brand J., Tieke M., et al. Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Adv. 2020; 4(13): 2967–78. https://doi.org/10.1182/bloodadvances.2020001640
- Panina I.Yu., Rumyantsev A.Sh., Menshutina M.A., Achkasova V.V., Degtereva O.A., Tugusheva F.A., et al. Specific functions of the endothelium in chronic kidney disease. literature review and personal data. Nefrologiya. 2007; 11(4): 28–46. https://elibrary.ru/jtygjh (in Russian)
- Kim K.S., Jung H., Shin I.K., Choi B.R., Kim D.H. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015; 87(7): 1104–12. https://doi.org/10.1002/jmv.24138
- Choreño-Parra J.A., Jiménez-Álvarez L.A., Cruz-Lagunas A., Rodríguez-Reyna T.S., Ramírez-Martínez G., Sandoval-Vega M., et al. Clinical and immunological factors that distinguish COVID-19 from pandemic influenza A(H1N1). Front. Immunol. 2021; 12: 593595. https://doi.org/10.3389/fimmu.2021.593595
- Sumikoshi M., Hashimoto K., Kawasaki Y., Sakuma H., Suzutani T., Suzuki H., et al. Human influenza virus infection and apoptosis induction in human vascular endothelial cells. J. Med. Virol. 2008; 80(6): 1072–8. https://doi.org/10.1002/jmv.21185
- Cassina A.M., Hodara R., Souza J.M., Thomson L., Castro L., Ischiropoulos H., et al. Cytochrome c nitration by peroxynitrite. J. Biol. Chem. 2000; 275(28): 21409–15. https://doi.org/10.1074/jbc.M909978199
- Halder U.C., Bagchi P., Chattopadhyay S., Dutta D., Chawla-Sarkar M. Cell death regulation during influenza A virus infection by matrix (M1) protein: a model of viral control over the cellular survival pathway. Cell Death Dis. 2011; 2(9): e197. https://doi.org/10.1038/cddis.2011.75
- Winn R.K., Harlan J.M. The role of endothelial cell apoptosis in inflammatory and immune diseases. J. Thromb. Haemost. 2005; 3(8): 1815–24. https://doi.org/10.1111/j.1538-7836.2005.01378.x
- Shevchenko YU.L., Stojko YU.M., Gudymovich V.G. Endothelium as a target of pathological effects of viral infection. Vestnik Natsional’nogo mediko-khirurgicheskogo tsentra im. N.I. Pirogova. 2022; 17(2): 11–6. https://doi.org/10.25881/20728255_2022_17_2_11 https://elibrary.ru/yzfzkv (in Russian)
- Gui R, Chen Q. Molecular events involved in influenza A virus-induced cell death. Front. Microbiol. 2022; 12: 797789. https://doi.org/10.3389/fmicb.2021.797789
- Zhirnov O.P., Konakova T.E., Wolff T., Klenk H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002; 76(4): 1617–25. https://doi.org/10.1128/jvi.76.4.1617-1625.2002
- Stasakova J., Ferko B., Kittel C., Sereinig S., Romanova J., Katinger H., et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005; 86(Pt. 1): 185–95. https://doi.org/10.1099/vir.0.80422-0
- Wang X., Zheng T., Lin L., Zhang Y., Peng X., Yan Y., et al. Influenza A virus induces autophagy by its hemagglutinin binding to cell surface heat shock protein 90AA1. Front. Microbiol. 2020; 11: 566348. https://doi.org/10.3389/fmicb.2020.566348
- Othumpangat S., Noti J.D., McMillen C.M., Beezhold D.H. ICAM-1 regulates the survival of influenza virus in lung epithelial cells during the early stages of infection. Virology. 2016; 487: 85–94. https://doi.org/10.1016/j.virol.2015.10.005
- Tinoco R., Deiro M., Lin M., Bradley L. P-selectin regulation of T cell immunity during influenza virus infection (49.14). J. Immunol. 2011; 186(1 Suppl.): 49.14. https://doi.org/10.4049/jimmunol.186.Supp.49.14
- Short K.R., Veldhuis Kroeze E.J., Reperant L.A., Richard M., Kuiken T. Influenza virus and endothelial cells: a species specific relationship. Front. Microbiol. 2014; 5: 653. https://doi.org/10.3389/fmicb.2014.00653
- Garcia C.C., Russo R.C., Guabiraba R., Fagundes C.T., Polidoro R.B., Tavares L.P., et al. Platelet-activating factor receptor plays a role in lung injury and death caused by Influenza A in mice. PLoS Pathog. 2010; 6(11): e1001171. https://doi.org/10.1371/journal.ppat.1001171
- Morichi S., Morishita N., Takeshita M., Ishida Y., Oana S., Yamanaka G., et al. Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) levels in the cerebrospinal fluid of children with influenza-associated encephalopathy. J. Infect. Chemother. 2017; 23(2): 80–4. https://doi.org/10.1016/j.jiac.2016.10.007
- Schmitz N., Kurrer M., Bachmann M.F., Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 2005; 79(10): 6441–8. https://doi.org/10.1128/JVI.79.10.6441-6448.2005
- Bawazeer A.O., Rosli S., Harpur C.M., Docherty C.A., Mansell A., Tate M.D. Interleukin-1β exacerbates disease and is a potential therapeutic target to reduce pulmonary inflammation during severe influenza A virus infection. Immunol. Cell Biol. 2021; 99(7): 737–48. https://doi.org/10.1111/imcb.12459
- Zhilinskaya I.N., Marchenko V.A., Kharchenko E.P. Comparison of fragments in human hemostatic proteins that mimics fragments in proteins of A/H1N1 viruses and coronaviruses. Molekulyarnaya genetika, mikrobiologiya i virusologiya. 2022; 40(4): 43–6. https://doi.org/10.17116/molgen20224004143 https://elibrary.ru/mwqoig (in Russian)
- Goldsteyn E.M. Influenza-associated mortality for circulatory and respiratory causes during the 2013-2014 through the 2018-2019 influenza seasons in Russia. Mezhdunarodnyi zhurnal prikladnykh i fundamental’nykh issledovanii. 2019; (12-1): 9–16. https://doi.org/10.17513/mjpfi.12945 https://elibrary.ru/dhthqt (in Russian)
Supplementary files

