



Активация и дисфункция эндотелия кровеносных сосудов при инфекции, вызванной вирусами гриппа типа А (Alphainfluenzavirus influenzae)
- Авторы: Марченко В.А.1, Жилинская И.Н.1
-
Учреждения:
- ФГБОУ ВО «Северо-Западный государственный медицинский университет имени И.И. Мечникова» Минздрава России
- Выпуск: Том 69, № 6 (2024)
- Страницы: 465-478
- Раздел: ОБЗОРЫ
- URL: https://virusjour.crie.ru/jour/article/view/16681
- DOI: https://doi.org/10.36233/0507-4088-264
- EDN: https://elibrary.ru/zujoza
- ID: 16681

Цитировать

Аннотация
Ежегодно тяжелое течение гриппа регистрируют у 3–5 млн больных, при этом у более 600 тыс. человек заболевание заканчивается летальным исходом. В основе тяжелых форм гриппа, как правило, лежит поражение эндотелия кровеносных сосудов. Так, вирусы гриппа А, включая подтипы A(H1N1)pdm09, A(H3N2), а также высокопатогенные вирусы гриппа птиц, способны инфицировать сосудистый эндотелий, приводя к активации и последующей дисфункции данных клеток. В свою очередь, эндотелиальная дисфункция характеризуется системным изменением морфофункциональных свойств клеток эндотелия, что приводит к нарушению тонуса кровеносных сосудов, тромбозу и другим осложнениям, а также одновременно является фактором риска и ключевым звеном патогенеза многих заболеваний сердечно-сосудистой системы. Таким образом, дисфункция эндотелия кровеносных сосудов является важным аспектом патогенеза тяжелого течения гриппа, что нужно учитывать в патогенетической терапии данного заболевания.
Цель данного обзора – проанализировать причины и уточнить механизмы развития активации и дисфункции эндотелия при инфекции, вызванной вирусами гриппа типа А.
Полный текст
Введение
Вирусы гриппа являются одними из наиболее распространенных возбудителей инфекционных заболеваний органов дыхания. Так, ежегодно гриппом болеет более 15% мирового населения, тогда как тяжелое течение гриппозной инфекции регистрируют у 3–5 млн больных [1]. В свою очередь, тяжелое течение гриппа нередко осложняется развитием геморрагического синдрома, что повышает риск возникновения геморрагического инсульта, инфаркта миокарда, острого коронарного синдрома, глубокого тромбоза вен и других сердечно-сосудистых осложнений [2‒5]. Согласно данным Всемирной организации здравоохранения, к группам риска по развитию тяжелых форм гриппа относят детей младше 6 лет, лиц старше 65 лет, беременных женщин, лиц с хронической соматической патологией, а также иммунокомпрометированных лиц.
Известно, что сосудистый эндотелий является мишенью для вирусов гриппа типа А (ВГА) [6‒8]. При тяжелом течении инфекции вирусы гриппа опосредуют чрезмерную активацию и поражение эндотелия кровеносных сосудов, что является причиной развития дисфункции эндотелия (ДЭ). В свою очередь, ДЭ является важным звеном патогенеза, а также фактором риска развития множества сердечно-сосудистой патологий.
В обзоре представлена актуальная информация, касающаяся механизмов активации и дисфункции эндотелия кровеносных сосудов при инфекции, вызванной ВГА.
Функции эндотелия кровеносных сосудов
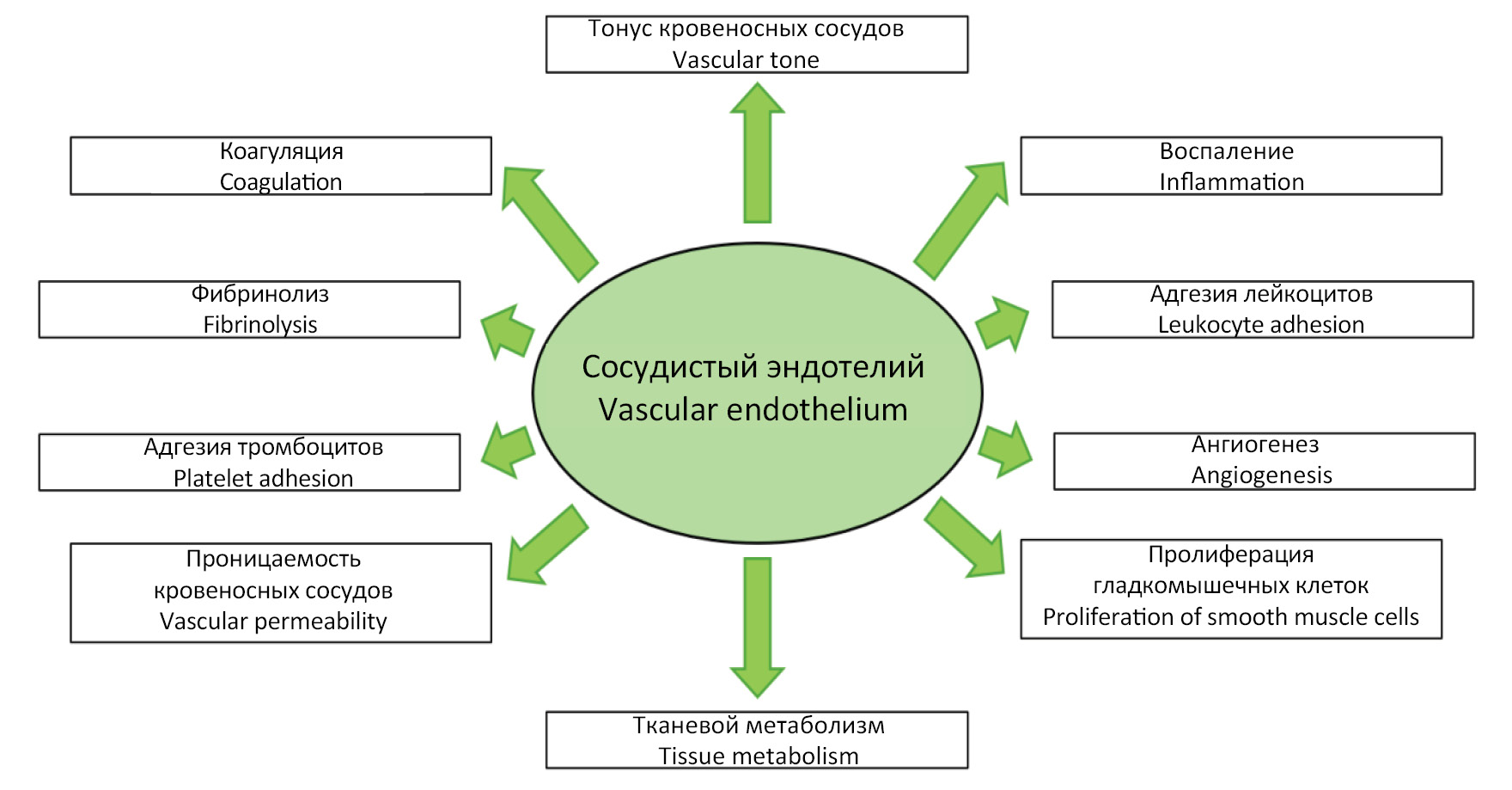
Эндотелий является гигантским «эндокринным» органом, распределенным по всем тканям организма человека, поддерживающим сосудистый и тканевой гомеостаз за счет продукции целого ряда биологически активных веществ. В физиологических условиях анатомически и функционально неповрежденный сосудистый эндотелий выполняет ряд функций, включая: 1) регуляцию тонуса кровеносных сосудов (вазомоторная функция); 2) регуляцию адгезии лейкоцитов (адгезивная функция); 3) регуляцию системы гемостаза (гемостатическая функция); 4) регуляцию ангиогенеза (ангиогенная функция); 5) регуляцию иммунных процессов (иммунная функция) и пр. (рис. 1) [9, 10].

Рис. 1. Функции эндотелия кровеносных сосудов.
Fig. 1. Vascular endothelium functions.
Эндотелий кровеносных сосудов как мишень для вирусов гриппа А
ВГА способны инфицировать различные клетки респираторного тракта, включая клетки мерцательного эпителия, а также бронхиолярные экзокриноциты [11, 12]. В качестве рецептора для адсорбции вирусы гриппа используют сиаловые кислоты. Для ВГА подтипов A(H1N1)pdm09 и A(H3N2) характерна высокая специфичность в отношении сиаловой кислоты с α-2,6-связью, которая преимущественно экспрессируется на эпителии верхних и нижних дыхательных путей, включая эпителий трахеи, бронхов, а также на альвеолоцитах 1-го типа [13‒15]. В ходе интенсивной репродукции вирусы сезонного гриппа оказывают прямое цитопатического действие на инфицированные эпителиоциты, что приводит к их значительному повреждению и гибели [16, 17]. Это позволяет вирусам проникать из входных ворот в регионарные кровеносные сосуды и взаимодействовать с сосудистым эндотелием.
В свою очередь, высокопатогенные вирусы гриппа птиц специфически связываются с α-2,3-сиаловой кислотой на поверхности клеток эпителия нижних дыхательных путей, включая эпителий бронхиол и альвеолоциты 2-го типа [18, 19]. Репликация вирусов гриппа в альвеолоцитах, как правило, приводит к апоптозу клеток, что позволяет вирусам контактировать с базолатеральной поверхностью эндотелиоцитов легочных капилляров [20, 21].
Установлено, что клетки эндотелия кровеносных сосудов, включая микрососуды легких, экспрессируют на своей поверхности оба типа сиаловых кислот (α-2,3 и α-2,6) [14, 22, 23]. Так, в исследованиях in vitro было показано, что клетки эндотелия легочных капилляров являются чувствительными в отношении ВГА, однако титр высокопатогенных вирусов гриппа A(H5N1) и A(H7N9) в культуре клеток эндотелия на несколько порядков выше, чем титр ВГА подтипов A(H1N1)pdm09 и A(H3N2) – 5–8 lg против 2,5–4 lg ТЦД50/мл [24‒26]. Эксперименты in vivo также подтверждают тот факт, что ВГА способны инфицировать клетки сосудистого эндотелия, а также что высокопатогенные вирусы гриппа птиц намного чаще поражают эндотелий кровеносных сосудов, что приводит к развитию таких жизнеугрожающих осложнений, как «цитокиновый шторм», острый респираторный дистресс-синдром (ОРДС) и синдром диссеминированного внутрисосудистого свертывания (ДВС-синдром) [27‒29]. Таким образом, клетки эндотелия являются не только чувствительными, но также пермиссивными в отношении высокопатогенных вирусов гриппа птиц и вирусов гриппа А(H1N1) и А(H3N2).
Активация и дисфункция эндотелия
Активация и дисфункция эндотелия являются близкими, но не тождественными понятиями [30]. Так, активацию эндотелия следует рассматривать как вариант ответа клеток на различные стимулы, интенсивность и/или длительность которых не превышает лимита клеточной адаптивной реакции. К таким активирующим стимулам можно отнести нарушение кровотока, цитокинемию, гипоксию, токсические вещества, патоген-ассоциированные молекулярные паттерны (PAMP), молекулярные паттерны, связанные с повреждением (DAMP), и пр. [31‒33]. Кроме того, активация эндотелия наблюдается при воздействии на клетки активных форм кислорода (АФК) и активных форм азота (АФА). Повышение уровня АФК, с одной стороны, является необходимым условием для реализации механизмов врожденного иммунитета, а с другой ‒ при несостоятельности системы антиоксидантной защиты вызывает оксидативный стресс и повреждение клеточных мембран [34].
Активация эндотелия представляет собой двухстадийный процесс. Так, в ходе первой стадии («активация I типа», или стимуляция эндотелия) эндотелиальные клетки практически мгновенно реагируют на стимул без изменения фенотипа и синтеза белков de novo [35]. Взаимодействие лиганда (например, гистамина) с рецепторами, связанными c G-белком, опосредует перестройку актинового цитоскелета, сокращение клеток и экзоцитоз из телец Вейбеля‒Паладе на поверхность клеток ряда белков, включая фактор Виллебранда (vWF) и P-селектин [36].
Вторая стадия («активация II типа») возникает как отсроченный ответ эндотелиальных клеток при воздействии стимула на протяжении нескольких часов или суток [30]. В основе активации эндотелия II типа лежит цитокин-опосредованная активация сигнального пути NF-κB, в результате чего возникает повышение экспрессии десятков генов и изменение фенотипа эндотелиальных клеток на «провоспалительный» [37, 38].
Активация эндотелия может носить как обратимый, так и необратимый характер. В том случае если воздействие триггерного фактора на эндотелий ограничено во времени, то гены, поддерживающие «провоспалительный» фенотип, постепенно подвергаются ингибированию, а экспрессия генов «вазопротективного» фенотипа восстанавливается [31]. В свою очередь, при чрезмерно выраженной и/или длительной активации эндотелия наблюдаются рецепторные, биохимические и морфоструктурные изменения, которые приводят к повреждению эндотелиоцитов и развитию ДЭ [39].
ДЭ характеризуется стойким нарушением морфофункциональных характеристик клеток эндотелия. При данном патологическом процессе наблюдается дисрегуляция процессов вазодилатации и вазоконстрикции, коагуляции и фибринолиза, ангиогенеза, воспаления и иммунного ответа. Именно ДЭ одновременно является фактором риска и основным звеном патогенеза многих заболеваний кардиоваскулярной системы, включая атеросклероз, артериальную гипертонию, ишемический инсульт и другие патологии [30, 40, 41]. Таким образом, грипп может инициировать развитие или отягощать течение заболеваний сердечно-сосудистой системы.
Активация и дисфункция клеток эндотелия при гриппе А: механизмы
На сегодняшний день многочисленные исследования in vitro и in vivo подтверждают тот факт, что ВГА способны вызывать не только активацию, но и дисфункцию клеток эндотелия кровеносных сосудов за счет дисрегуляции многочисленных клеточных процессов, в результате изменения экспрессии более чем 100 генов-мишеней [42]. Ниже представлены механизмы развития ДЭ при гриппе.
Оксидативный стресс и снижение биодоступности оксида азота (NO)
«Краеугольным камнем» эндотелиальной дисфункции кровеносных сосудов принято считать снижение синтеза и/или биодоступности NO в результате дисрегуляции экспрессии и/или активности эндотелиальной синтазы оксида азота (eNOS) [43]. В физиологических условиях eNOS постоянно экспрессируется в сосудистом эндотелии и генерирует NO в низких концентрациях. Это чрезвычайно важно, т.к. именно в низкой концентрации NO обладает противовоспалительным, антипролиферативным, антитромбогенным и вазодилатирующим эффектом [31].
При инфицировании клеток эндотелия ВГА отмечается изменение экспрессии данного эндотелиального фактора. Так, вирус гриппа A(H1N1)pdm09 значительно снижает уровень экспрессии eNOS в культуре эндотелиальных клеток, а также вызывает длительное и системное снижение экспрессии данного фермента in vivo (не менее 2 мес) [8, 44]. Полученные результаты согласуются с эпидемиологическими данными о положительной корреляции между заболеваемостью гриппом и смертностью от сердечно-сосудистых заболеваний в течение 2 мес после окончания эпидемии (так называемая «дополнительная» смертность) [45].
Установлено, что одной из основных причин дисрегуляции экспрессии eNOS, вызванной ВГА, является свободнорадикальное повреждение клеток в результате оксидативного стресса. При активации эндотелия в клетках происходит избыточное образование АФК митохондриями. При несостоятельности антиоксидантной системы свободные радикалы, в частности супероксид анион-радикал (О2•‒), способен связываться с NO с образованием пероксинитрита (ONOO−), обладающего крайне высоким окислительным потенциалом [46]. В свою очередь, пероксинитрит вызывает окисление тетрогидробиоптерина – одного из кофакторов eNOS, что ведет к разобщению eNOS со своим субстратом, в результате чего данный фермент начинает производить супероксид анион-радикал вместо NO [47]. Таким образом, концентрация АФК и АФА в клетке резко повышается, что усиливает повреждение эндотелиальных клеток и отягощает ДЭ.
Следует отметить, что компенсаторные механизмы, направленные на восстановление биодоступности NO в условиях тяжело протекающего инфекционного процесса, как правило, являются несостоятельными. Так, при выраженной воспалительной реакции клетки эндотелия, а также макрофаги/моноциты начинают синтезировать индуцибельную синтазу оксида азота (iNOS) [48]. Однако данная изоформа фермента генерирует значительно более высокие концентрации NO, что обуславливает преобладание непрямых эффектов, связанных с образованием пероксинитрита и свободно-радикальными повреждением клеток [49].
Цитокинемия и «цитокиновый шторм»
На примере высокопатогенного вируса гриппа A(H5N1) было показано, что при инфицировании клеток эндотелия микрососудов легких наблюдается как более выраженная активация фактора транскрипции NF-κB, так и дополнительная активация сигнальных путей митоген-активируемой протеинкиназы (MAPK) [50]. В частности, высокопатогенные вирусы гриппа активируют p38 MAPK-путь, что проявляется в чрезмерном и неконтролируемом синтезе провоспалительных цитокинов. Важно отметить, что в этом случае именно активированный эндотелий микрососудов легких становится основным продуцентом провоспалительных цитокинов (интерлейкинов (IL) 1β, IL-6, фактора некроза опухоли-альфа (TNF-α)) и хемокинов (CXCL10, RANTES) [42, 51, 52]. Кроме того, возникающая гиперцитокинемия опосредует системное повреждение эндотелия других сосудистых регионов, что является причиной развития «цитокинового шторма» и ОРДС.
Необходимо подчеркнуть, что важную роль в развитии «цитокинового шторма» при гриппе играет фактор транскрипции KLF2. В физиологических условиях данный транскрипционный фактор поддерживает биодоступность NO на оптимальном уровне, а также барьерные свойства и тромборезистентность клеток эндотелия [53‒55]. Результаты исследования R. Huang и соавт. показали, что при инфицировании высокопатогенными вирусами гриппа мышей Balb/C уровень экспрессии фактора транскрипции KLF2 в эндотелии микрососудов легких значительно снижен, что обуславливает развитие «цитокинового шторма», острого повреждения легких и ОРДС. Эти изменения соотносятся с тем, что при чрезмерной активации фактора транскрипции NF-κB, наблюдается снижение активности фактора KLF2 [56].
Нарушение метаболических процессов
ВГА, включая подтипы A(H1N1)pdm09, A(H3N2) и A(H5N1), угнетают метаболические процессы в клетках эндотелия и вызывают двукратное снижение дегидрогеназной активности. Следует отметить, что снижение активности дегидрогеназ, но в меньшей степени, также наблюдается и в ответ на внесение в культуральную среду отдельных поверхностных белков разных подтипов ВГА – гемагглютинина и нейраминидазы [57].
Вероятно, одной из причин столь выраженных изменений метаболических процессов является воздействие на клетки эндотелия провоспалительных цитокинов, например TNF-α, IL-1β и IL-6, способных подавлять активность митохондрий и, как следствие, катаболические процессы [58]. Кроме того, образование внутриклеточного пероксинитрита на фоне дисрегуляции экспрессии eNOS обуславливает повреждение мембран митохондрий, что также способствует снижению митохондриальной активности.
Нарушение барьерных свойств, изменение морфологии и повышение проницаемости эндотелия
Важное значение в поддержании барьерных свойств эндотелия отводят интактному гликокаликсу. В состав гликокаликса сосудистого эндотелия в основном входят протеогликаны, а также гликопротеины, связанные с сиаловыми кислотами [59]. Ферментативная активность нейраминидазы вируса гриппа позволяет расщеплять связь между терминальным остатком сиаловой кислоты и гликопротеином в составе гликокаликса эндотелиоцитов, что обуславливает его истончение и отслоение. Кроме того, активированный ВГА эндотелий начинает синтезировать матриксные металлопротеиназы-2 и -9, которые усиливают повреждение гликокаликса. В итоге эндотелиальный гликокаликс подвергается деградации, теряет свой отрицательный заряд, в результате чего эндотелий становятся проницаем для молекул с относительно высокой молекулярной массой [60].
Проницаемость эндотелия также зависит от целостности межклеточных контактов (плотных, адгезивных и щелевидных) [61]. В исследованиях in vitro было показано, что ВГА вызывают деградацию различных белков, входящих в состав межклеточных контактов (β-катенина, клаудина-5, VE-кадгерина, белка ZO-1 и коннексинов), а также реорганизацию актинового цитоскелета эндотелиоцитов с образованием стресс-индуцированных фибрилл [58, 62]. Вследствие этого клетки эндотелия сокращаются и округляются с образованием промежутков между ними, что является причиной увеличения сосудистой проницаемости [42].
Следует отметить, что повреждение эндотелиального гликокаликса, а также деградация белков, входящих в состав межклеточных контактов, может наблюдаться при воздействии на клетки ндотелия высокими концентрациями цитокинов (IL-1β и TNF-α), что также значительно повышает проницаемость эндотелия микрососудов легких и стоит в основе развития острого поражения легких и ОРДС [63‒66].
Нарушение адгезивных свойств
Активация сигнального пути NF-κB при гриппе приводит к повышению экспрессии различных молекул клеточной адгезии, включая адгезивные рецепторы суперсемейства иммуноглобулинов (ICAM-1, ICAM-2, VCAM-1, PECAM-1) и селектины (P-селектин, E-селектин) [20, 67‒69].
В свою очередь, повышение адгезивных свойств позволяет циркулирующим лейкоцитам осуществлять адгезию на клетках эндотелия, а затем мигрировать из сосудистого русла в очаг воспаления. Однако при системной воспалительной реакции на фоне тяжелого гриппа возникает выраженная и нерегулируемая адгезия лейкоцитов к эндотелию кровеносных сосудов, причем тяжесть течения гриппа во многом зависит от степени вовлечения в воспалительную реакцию нейтрофилов [70]. Именно нейтрофилы при тяжелом гриппе становятся одними из основных продуцентов АФК и медиаторов воспаления, а также синтезируют внеклеточные нейтрофильные ловушки (NETs), опосредуя повреждение клеток [71‒73].
Нарушение гемостаза и фибринолиза
Для тяжелого течения гриппа характерны нарушения процессов коагуляции и фибринолиза по типу ДВС-синдрома [29]. ВГА опосредуют активацию как внешнего, так и внутреннего пути коагуляции (рис. 2). В ответ на инфицирование или в результате воздействия на клетки эндотелия высоких концентраций провоспалительных цитокинов (TNF и IL-1β) в клетках повышается экспрессия эндотелиальных факторов с прокоагуляторными свойствами: vWF, фактора активации тромбоцитов (PAF), ингибитора активатора плазминогена-1 (PAI-1), а также тканевого фактора (TF) – ключевого гликопротеина, запускающего внешней путь свертывания [20, 74‒77]. В своих исследования F. Visseren и соавт. показали, что инфицирование клеток эндотелия вирусом гриппа A(H1N1) и A(H3N2) увеличивает экспрессию TF, что снижает время свертывания крови на 55% в течение 3 ч после инфицирования [78]. Аналогичные данные по увеличению концентрации TF в бронхоальвеолярном лаваже были получены при инфицировании мышей Balb/C вирусом A(H1N1) [79].

Рис. 2. Каскад коагуляции.
1 ‒ интактные эндотелиальные клетки (ЭК) обеспечивают тромборезистентность за счет постоянного синтеза ряда антикоагулянтов (тромбомодулина, антитромбина, ингибитора пути тканевого активатора и АДФазы); 2 ‒ каскад коагуляции, как правило, возникает вследствие повреждения ЭК и связан с активацией факторов свертывания крови при контакте с тканевым фактором и коллагеном из субэндотелиальной слоя, а также с высвобождением из клеток фактора Виллебранда (vWF); 3 ‒ активация тромбоцитов возникает при взаимодействии с тканевым фактором, коллагеном и vWF. Активированные тромбоциты высвобождают ряд медиаторов, таких как АДФ и vWF, что приводит к дальнейшему рекрутингу, активации и агрегации тромбоцитов с образованием первичного тромбоцитарного сгустка (первичный гемостаз); 4 ‒ внешний путь свертывания инициируется при контакте фактора VII с тканевым фактором; 5 ‒ внутренний путь инициируется при контакте фактора XII с коллагеном; 6 ‒ внешний и внутренний пути приводят к инициированию общего пути свертывания, который содержит каскады, участвующие в активации фактора X и тромбина с образованием нитей фибрина; 7 ‒ нити фибрина способствуют повышению стабильности тромбоцитарного сгустка и приводят к образованию тромбоцитарно-фибринового сгустка (вторичный гемостаз); 8 ‒ калликреин, тканевой активатор плазминогена (tPA) или урокиназный активатор плазминогена (uPA) конвертируют плазминоген в плазмин, который затем разрушает и реабсорбирует полимеризованные нити фибрина, что необходимо для разрушения сгустков в рамках процесса фибринолиза. Подчеркнуты эндотелиальные факторы, концентрация в крови которых при гриппозной инфекции достоверно изменяется (см. таблицу).
Fig. 2. The coagulation cascades.
1 ‒ intact endothelial cells express antiplatelet and anticoagulant agents (thrombomodulin, antithrombin, tissue factor pathway inhibitor and ADPase) that prevent aggregation of platelet and fibrin formation; 2 ‒ coagulation is usually initiated by an injury to the endothelium, with the exposure of tissue factor and collagen from the subendothelium to the blood factors and the release of von Willebrand factor (vWF); 3 ‒ activation of platelets is initiated by exposure to tissue factor, collagen and vWF. Activated platelets release several mediators (including ADP and vWF), leading to further platelet recruitment, activation, aggregation and plug formation (primary hemostasis); 4 ‒ the extrinsic pathway is initiated by the interaction between tissue factor and Factor VII; 5 ‒ the intrinsic pathway is initiated by the exposure of collagen to Factor XII; 6 ‒ the extrinsic and intrinsic coagulation pathways lead into the final common pathway, which contains cascades involved in the production of thrombin, activated Factor X and the formation of fibrin strands; 7 ‒ fibrin strands increase stability of the platelet plug and lead to the formation of platelet-fibrin clot (secondary hemostasis); 8 ‒ kallikrein, tissue plasminogen activator (tPA) or urokinase plasminogen activator (uPA) convert plasminogen to plasmin, which then degrades and reabsorbs the fibrin strands in process called fibrinolysis. Endothelial factors whose concentration in influenza A virus infection is reliably changed are underlined (see Table).
Следует отметить важную роль в патогенезе гриппа фактора PAI-1. Данный эндотелиальный фактор является антагонистом двух белков системы фибринолиза – урокиназного и TF плазминогена (uPA и tPA), которые осуществляют расщепление плазминогена до плазмина [80]. В свою очередь, плазмин может быть использован вирусами гриппа для гидролиза белка ‒ предшественника гемагглютинина (HA0), что необходимо для созревания вновь синтезированных вирионов. Таким образом, повышение экспрессии PAI-1, с одной стороны, снижает инфекционную активность вируса, а с другой ‒ может становиться причиной ингибирования фибринолиза и усиления процессов тромбообразования при гриппе.
Активация внешнего и внутреннего пути in vivo возникает при повреждении клеток эндотелия. В частности, расположенные субэндотелиально TF и коллаген в результате десквамации эндотелия начинают взаимодействовать с факторами VII и XII, что необходимо для активации общего пути коагуляции с активацией тромбоцитов и образованием тромбоцитарных сгустков [29]. Таким образом, степень поражения эндотелия сосудов вероятно коррелирует с выраженностью активации системы коагуляции при гриппе.
Помимо участия в гемостазе, тромбоциты также играют важную роль в иммунном ответе. Так, тромбоциты способны захватывать и секвестрировать вирусы, что позволяет ограничивать их распространение [81]. Однако, вероятно, эндоцитоз вирусов гриппа не всегда носит завершенный характер. В своих исследованиях M. Koupenova и соавт. обнаружили, что часть фрагментов вирусных частиц остаются экспонированными на поверхности тромбоцитов. Это, в свою очередь, может опосредовать более длительный контакт клеток сосудистого эндотелия с патоген-ассоциированными молекулярными паттернами вируса, приводя к более выраженной активации эндотелиоцитов.
Истощение прокоагулятных факторов и развивающаяся тромбоцитопения могут в последующем приводить к фазе гипокоагуляции [82]. Вирусы гриппа за счет нейраминидазной активности могут опосредовать отщепление сиаловых кислот от гликокаликса тромбоцитов, что усиливает их удаление из кровотока вплоть до развития тромбоцитопении [83]. Кроме того, выраженность тромбоцитопении зависит от подтипа вируса. Так, вирус гриппа A(H5N1) вызывает более выраженную тромбоцитопению по сравнению с ВГА подтипов A(H1N1) и А(H3N2).
Нарушение иммунных процессов
Инфицированный сосудистый эндотелий не способен адекватно поддерживать иммунные процессы, в первую очередь связанные с представлением антигенов иммунокомпетентным клеткам. Кроме того, также наблюдается дисрегуляция в активации клеток врожденного иммунитета [84]. Как уже было отмечено ранее, в клетках эндотелия, инфицированных ВГА, значительно повышается синтез IL-1β [85]. Этот провоспалительный цитокин играет важную роль во врожденном иммунном ответе, т.к. опосредует процесс антиген-зависимой дифференцировки Т-лимфоцитов, а также дифференциацию дендритных клеток. Таким образом, при тяжелом гриппе с вовлечением в воспалительный процесс клеток сосудистого эндотелия за счет синтеза IL-1β происходит избыточная инфильтрация легочной паренхимы иммунными клетками, что может обуславливать острое повреждение легких.
Нарушение процессов ангиогенеза
Изменение уровня экспрессии эндотелиальных факторов, влияющих на ангиогенез, при гриппозной инфекции не характерно [86]. Однако результаты исследования S. Morichi и соавт. показали, что у детей при развитии вирус-индуцированной энцефалопатии в спинномозговой жидкости отмечают повышение экспрессии двух маркеров ангиогенеза – фактора роста эндотелия сосудов (VEGF-A) и тромбоцитарного фактора роста (PDGF).
Апоптоз
При инфицировании эндотелиоцитов ВГА апоптоз клеток может возникать за счет активации как рецептор-зависимого (внешнего), так и митохондриального (внутреннего) сигнальных путей. Одной из причин активации внешнего пути апоптоза является воздействие на клетку высоких концентраций TNF-α. Это подтверждается тем, что в инфицированных клетках эндотелия повышается экспрессия каспазы-3 – эффекторной протеазы, расщепляющей цитоскелет и активирующей эндонуклеазу на терминальных стадиях апоптоза [26, 87].
Внутренний путь активации апоптоза реализуется при повреждении мембраны митохондрий. Внутриклеточные активные формы кислорода и азота, в частности пероксинитрит, опосредуют повреждение мембран данных органелл с высвобождением цитохрома с [88]. Кроме того, основной белок вируса гриппа М1 способен связываться с важным компонентом цитопротекции – белком теплового шока 70 (Hsp70), в результате чего происходит диссоциация связи Hsp70 и белка APAF-1 с высвобождением последнего и образованием апоптосом [89]. В итоге вирус-индуцируемый апоптоз приводит к нарушению целостности эндотелиального барьера, что становится причиной повышения сосудистой проницаемости, а также играет ключевую роль в патогенезе тромбоза, ДВС-синдрома, васкулита и атеросклероза [62, 90, 91].
Следует отметить, что при вирусных инфекциях ранняя активация апоптоза в большинстве случаев позволяет значительно подавить репликацию вируса, тогда как задержка апоптоза инфицированных клеток становится причиной распространения вирусных частиц в организме [92]. В свою очередь, в отношении вируса гриппа в исследованиях in vitro были получены данные о том, что апоптоз инфицированных клеток является отсроченным ввиду антиапоптической активности вирусного белка NS1 [93, 94]. Кроме того, помимо апоптоза ВГА способен индуцировать другие программируемые варианты гибели клетки, включая: 1) некроптоз (за счет белков HA, NS1, PB1); 2) пироптоз (за счет белков М2, PB1-F2, PB2); 3) аутофагию (за счет белка HA) [92, 95].
Исходя из вышеизложенного становится очевидным, что при тяжелом течении гриппозной инфекции, вызванной ВГА, наблюдается нарушение всех функций эндотелия: вазомоторной, адгезивной, гемостатической, ангиогенной, иммунной и др. (таблица). В качестве основных причин развития ДЭ вероятно следует рассматривать воздействие на эндотелий сразу нескольких флогогенных факторов, среди которых наибольшее значение имеют цитопатическое действие вируса, оксидативный стресс, цитокинемия и гипоксия. Кроме того, не только сам вирус, но и его белки (например, гемагглютинин и нейраминидаза) способны вызывать выраженную активацию эндотелиоцитов, что может становиться причиной развития их дисфункции [57].
Таблица. Нарушения функций сосудистого эндотелия, вызванные вирусами гриппа типа А
Table. Alteration of vascular endothelium functions caused by Influenza A Viruses
Функции клеток эндотелия Functions of endothelial cells | Эндотелиальные факторы Endothelial factors | Источник Reference |
Вазомоторная Vasomotor | NO (за счет eNOS) ↓ NO (by eNOS) ↓ | |
Адгезивная Adhesion | ICAM-1 ↑ | |
P-селектин/P-selectin ↑ | ||
E-селектин/E-selectin ↑ | [98] | |
PECAM-1 ↑ | ||
Гемостатическая Hemostatic | tPA ↔ / ↑ | |
PAI-1 ↔ / ↑ | ||
vWF ↑ | [20] | |
PAF ↑ | [99] | |
TF ↑ | ||
Ангиогенная Angiogenic | VEFG ↑ PDGF ↑ | [100] |
Иммунная Immune | IL-1 ↑ |
Примечание. ↔ – модуляция экспрессии; ↑ – повышение экспрессии; ↓ – снижение экспрессии. ICAM-1 – межклеточная молекула адгезии-1; PECAM-1 – молекула адгезии тромбоцитов и эндотелиоцитов-1; tPA – тканевой активатор плазминогена; PAI-1 – ингибитор активатора плазминогена-1; vWF – фактор Виллебранда; PAF – фактор активации тромбоцитов; TF – тканевой фактор; VEFG – фактор роста эндотелия сосудов; PDGF – фактор роста тромбоцитов; IL-1 – интерлейкин-1.
Note. ↔ – modulation of the expression; ↑ – increased expression; ↓ – decreased expression. ICAM-1 – Intercellular adhesion molecule-1; PECAM-1 – platelet and endothelial cell adhesion molecule-1; tPA – tissue plasminogen activator; PAI-1 – plasminogen activator inhibitor-1; vWF – von Willebrand factor; PAF – platelet-activating factor; TF – tissue factor; VEFG – vascular endothelial growth factor; PDGF – platelet-derived growth factor; IL-1 – Interleukin-1.
Молекулярная мимикрия
Можно предположить, что вероятной причиной развития ДЭ при гриппе также является молекулярная мимикрия. Так, в составе разных штаммов вирусов гриппа A(H1N1)pdm09, а также A(H3N2) (данные не опубликованы) обнаружено множество аминокислотных последовательностей в белках с высокой степенью гомологии c аминокислотными последовательностями в различных белках системы гемостаза и эндотелиальных факторах, включая vWF, eNOS, PAI-1, TF, tPA, факторах свертывания крови (III, V, VI, VII, VIII, IX, X, XI, XIII) и др. В частности, в вышеперечисленных белках и различных белках вирусов гриппа обнаружены многочисленные последовательности длиной 12 аминокислотных остатков с гомологией, превышающей 80% [103].
Биоинформационный анализ позволил обнаружить в составе вируса гриппа A/H1N1, вызвавшего пандемию 1918–1920 гг., ряд уникальных последовательностей, мимикрирующих фрагменты в составе белков системы гемостаза и фибринолиза (фибриногена, TF, антитромбина-III, протромбина, плазминогена, урокиназного активатора плазминогена и др.), которые отсутствуют в вирусе гриппа A(H1N1)pdm09, выделенного в 2016 г. [92].
Интересно, что при сравнении современных циркулирующих штаммов вирусов гриппа A(H1N1)pdm09 и A(H3N2) в составе последних имеется большое количество аминокислотных последовательностей, гомологичных последовательностям различных факторов системы фибринолиза (α2-антиплазмина, α2-макроглобулина, тромбомодулина, урокиназного активатора плазминогена, калликреина). Стоит отметить, что консервативность данных последовательностей как по месторасположению, так и по аминокислотному составу сохраняется в белках вирусов A(H3N2) на протяжении 50 лет. Вероятно, данная особенность позволяет вирусам гриппа A(H3N2) вызывать более интенсивный эпидемический процесс (по сравнению с вирусом A(H1N1)pdm09) и обуславливать более высокий уровень дополнительной смертности от гриппа у больных с сопутствующими сердечно-сосудистыми заболеваниями [104]. Таким образом, наличие мимикрирующих последовательностей в белках системы гемостаза и вирусов гриппа, по всей видимости, повышает их вирулентность, т.к. позволяет нарушать процессы коагуляции и фибринолиза.
Известно, что тяжесть ДЭ при гриппе зависит не только от вируса (вирулентности штамма, инфицирующей дозы) и резистентности организма (предсуществующего иммунитета, генетической предрасположенности к развитию тяжелых форм заболевания), но и от состояния сердечно-сосудистой системы. Так, наличие острых и хронических заболеваний кардиоваскулярной системы у больных обуславливает более тяжелое течение гриппа, что связанно с вовлечением эндотелия кровеносных сосудов в патологический процесс. Это подтверждается тем, что после окончания эпидемии у пациентов с сердечно-сосудистыми заболеваниями показатель дополнительной смертности от гриппа составляет 481 на 100 тыс. населения против 2 на 100 тыс. населения среди здоровых взрослых без соматических заболеваний.
Заключение
ВГА способны вызывать активацию и дисфункцию эндотелия кровеносных сосудов, что является ключевым звеном в патогенезе тяжелого гриппа, обуславливая развитие осложнений в остром и отсроченном периоде инфекции. В свою очередь, ДЭ одновременно является фактором риска и основным звеном патогенеза многих заболеваний сердечно-сосудистой системы, включая атеросклероз, артериальную гипертонию, инфаркт миокарда, ишемический инсульт и прочие патологии.
Несмотря на наличие вакцинации как основной меры профилактики гриппа и ряда этиотропных препаратов из разных клинико-фармакологических групп, эффективно ингибирующих различные этапы репродукции вируса, важным направлением остается оптимизация схем патогенетической терапии гриппа. Учитывая высокий риск развития тяжелого течения гриппа у пациентов из групп риска, особенно у лиц старше 65 лет с хроническими заболеваниями сердечно-сосудистой системы, в рамках патогенетической терапии целесообразным является назначение химиопрепаратов с проверенной эндотелиопротективной активностью с целью коррекции эндотелиальной дисфункции.
Об авторах
Владимир Александрович Марченко
ФГБОУ ВО «Северо-Западный государственный медицинский университет имени И.И. Мечникова» Минздрава России
Автор, ответственный за переписку.
Email: vmarcenco@mail.ru
ORCID iD: 0000-0001-6870-3157
канд. мед. наук, доцент кафедры медицинской микробиологии
Россия, 191015, г. Санкт-ПетербургИрина Николаевна Жилинская
ФГБОУ ВО «Северо-Западный государственный медицинский университет имени И.И. Мечникова» Минздрава России
Email: vmarcenco@mail.ru
ORCID iD: 0000-0002-0084-1323
д-р биол. наук, профессор кафедры медицинской микробиологии
Россия, 191015, г. Санкт-ПетербургСписок литературы
- Office WHOEMR. Global Influenza Strategy 2019–2030. Weekly Epidemiological Record; 2019.
- Boehme A.K., Luna J., Kulick E.R., Kamel H., Elkind M.S.V. Influenza-like illness as a trigger for ischemic stroke. Ann. Clin. Transl. Neurol. 2018; 5(4): 45663. https://doi.org/10.1002/acn3.545
- Muscente F., De Caterina R. Causal relationship between influenza infection and risk of acute myocardial infarction: pathophysiological hypothesis and clinical implications. Eur. Heart J. 2020; 22(Suppl. E): E68–72. https://doi.org/10.1093/eurheartj/suaa064
- Skaarup K.G., Modin D., Nielsen L., Jensen J.U.S., Biering-Sørensen T. Influenza and cardiovascular disease pathophysiology: strings attached. Eur. Heart J. 2023;25(Suppl. A): A5–11. https://doi.org/10.1093/eurheartjsupp/suac117
- Rubino R., Imburgia C., Bonura S., Trizzino M., Iaria C., Cascio A. Thromboembolic events in patients with influenza: a scoping review. Viruses. 2022; 14(12): 2817. https://doi.org/10.3390/v14122817
- Short K.R., Kuiken T., Van Riel D. Role of endothelial cells in the pathogenesis of influenza in humans. J. Infect. Dis. 2019; 220(11): 1859–60. https://doi.org/10.1093/infdis/jiz349
- Armstrong S.M., Darwish I., Lee W.L. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence. 2013; 4(6): 537–42. https://doi.org/10.4161/viru.25779
- Марченко В.А., Барашкова С.В., Зелинская И.А., Торопова Я.Г., Рэмзи Э.С., Жилинская И.Н. Экспрессия эндотелиальных факторов в клетках эндотелия человека при инфекции, вызванной вирусом гриппа А(H1N1)pdm09 (Orthomyxoviridae; Alphainfluenzavirus). Вопросы вирусологии. 2021; 66(3): 198–210. https://doi.org/10.36233/0507-4088-48 https://elibrary.ru/wsxlvb
- Aird W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007; 100(2): 158–73. https://doi.org/10.1161/01.RES.0000255691.76142.4a
- Zhang J., Defelice A.F., Hanig J.P., Colatsky T. Biomarkers of endothelial cell activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol. Pathol. 2010; 38(6): 856–71. https://doi.org/10.1177/0192623310378866
- Matrosovich M.N., Matrosovich T.Y., Gray T., Roberts N.A., Klenk H.D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA. 2004; 101(13): 4620–4. https://doi.org/10.1073/pnas.0308001101
- Ibricevic A., Pekosz A., Walter M.J., Newby C., Battaile J.T., Brown E.G., et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006; 80(15): 7469–80. https://doi.org/10.1128/JVI.02677-05
- Abe Y., Smith C.W., Katkin J.P., Thurmon L.M., Xu X., Mendoza L.H., et al. Endothelial alpha 2,6-linked sialic acid inhibits VCAM-1-dependent adhesion under flow conditions. J. Immunol. 1999; 163(5): 2867–76.
- Cioffi D.L., Pandey S., Alvare D.F., Cioffi E.A. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell Mol. Physiol. 2012; 302(10): L1067–77. https://doi.org/10.1152/ajplung.00190.2011
- Denney L., Ho L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018; 41(4): 218–33. https://doi.org/10.1016/j.bj.2018.08.004
- Herold S., Becker C., Ridge K.M., Budinger G.R. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur. Respir. J. 2015; 45(5): 1463–78. https://doi.org/10.1183/09031936.00186214
- Herold S., Steinmueller M., von Wulffen W., Cakarova L., Pinto R., Pleschka S., et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008; 205(13): 3065–77. https://doi.org/10.1084/jem.20080201
- Zeng H., Goldsmith C.S., Maines T.R., Belser J.A., Gustin K.M., Pekosz A., et al. Tropism and infectivity of influenza virus, including highly pathogenic avian H5N1 virus, in ferret tracheal differentiated primary epithelial cell cultures. J. Virol. 2013; 87(5): 2597–607. https://doi.org/10.1128/JVI.02885-12
- Kumlin U., Olofsson S., Dimock K., Arnberg N. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir. Viruses. 2008; 2(5): 147–54. https://doi.org/10.1111/j.1750-2659.2008.00051.x
- Sugiyama M.G., Gamage A., Zyla R., Armstrong S.M., Advani S., Advani A., et al. Influenza virus infection induces platelet-endothelial adhesion which contributes to lung injury. J. Virol. 2015; 90(4): 1812–23. https://doi.org/10.1128/JVI.02599-15
- Lee S., Hirohama M., Noguchi M., Nagata K., Kawaguchi A. Influenza A virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the type I interferon signaling pathway in a mutually exclusive manner. J. Virol. 2018; 92(14): e00396-18. https://doi.org/10.1128/JVI.00396-18
- Chan M.C., Chan R.W., Yu W.C., Ho C.C., Chui W.H., Lo C.K., et al. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir. Res. 2009; 10(1): 102. https://doi.org/10.1186/1465-9921-10-102
- Zeng H., Pappas C., Belser J.A., Houser K.V., Zhong W., Wadford D.A., et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J. Virol. 2012; 86(2): 667–78. https://doi.org/10.1128/JVI.06348-11
- Chan L.L.Y., Hui K.P.Y., Kuok D.I.T., Bui C.H.T., Ng K.C., Mok C.K.P., et al. Risk assessment of the tropism and pathogenesis of the highly pathogenic avian influenza A/H7N9 virus using ex vivo and in vitro cultures of human respiratory tract. J. Infect. Dis. 2019; 220(4): 578–88. https://doi.org/10.1093/infdis/jiz165
- Simon P., de La Vega M.A., Paradis É., Mendoza E., Coombs K.M., Kobasa D., et al. Avian influenza viruses that cause highly virulent infections in humans exhibit distinct replicative properties in contrast to human H1N1 viruses. Sci. Rep. 2016; 6: 24154. https://doi.org/10.1038/srep24154
- Han T., Lai Y., Jiang Y., Liu X., Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading to microvascular leakage and release of pro-inflammatory cytokines. PeerJ. 2021; 9: e11892. https://doi.org/10.7717/peerj.11892
- Gu Y., Zuo X., Zhang S., Ouyang Z., Jiang S., Wang F., et al. The mechanism behind influenza virus cytokine storm. Viruses. 2021; 13(7): 1362. https://doi.org/10.3390/v13071362
- Tang B.M., Cootes T., McLean A.S. From influenza-induced acute lung injury to multiorgan failure. In: Annual Update in Intensive Care and Emergency Medicine 2019. 2018: 449–58. https://doi.org/10.1007/978-3-030-06067-1_35
- Yang Y., Tang H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016; 13(4): 432–42. https://doi.org/10.1038/cmi.2016.1
- Zhang J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc. Med. 2022; 23(2): 73. https://doi.org/10.31083/j.rcm2302073
- Immanuel J., Yun S. Vascular inflammatory diseases and endothelial phenotypes. Cells. 2023; 12(12): 1640. https://doi.org/10.3390/cells12121640
- Мельникова Ю.С., Макарова Т.П. Эндотелиальная дисфункция как центральное звено патогенеза хронических болезней. Казанский медицинский журнал. 2015; 96(4): 659–65. https://doi.org/10.17750/KMJ2015-659 https://elibrary.ru/ubegwv
- Власова Т.И., Петрищев Н.Н., Власов Т.Д. Дисфункция эндотелия как типовое патологическое состояние. Регионарное кровообращение и микроциркуляция. 2022; 21(2): 4–15. https://doi.org/10.24884/1682-6655-2022-21-2-4-15 https://elibrary.ru/zheshs
- Yang Y., Bazhin A.V., Werner J., Karakhanova S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013; 32(3): 249–70. https://doi.org/10.3109/08830185.2012.755176
- Bach F.H., Robson S.C., Ferran C., Winkler H., Millan M.T., Stuhlmeier K.M., et al. Endothelial cell activation and thromboregulation during xenograft rejection. Immunol. Rev. 1994; 141: 5–30. https://doi.org/10.1111/j.1600-065x.1994.tb00870.x
- Pober J.S., Sessa W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007; 7(10): 803–15. https://doi.org/10.1038/nri2171
- Бигильдеев А.Е., Чепурных Ю.Ф., Петинати Н.А., Дризе Н.И. Особенности экспрессии генов сигнального пути NF-kB в тканях облученных мышей и у старых животных. Радиационная биология. Радиоэкология. 2019; 59(6): 565–74. https://doi.org/10.1134/S0869803119060031 https://elibrary.ru/ebdunp
- Waitkus M.S., Harris D.P., DiCorleto P.E. Mechanisms of Endothelial Activation. In: Mackay I.R., Rose N.R., Diamond B., Davidson A., eds. Encyclopedia of Medical Immunology. New York: Springer; 2014. https://doi.org/10.1007/978-0-387-84828-0_183
- Endemann D.H., Schiffrin E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004; 15(8): 1983–92. https://doi.org/10.1097/01.ASN.0000132474.50966.DA
- Hadi H.A., Carr C.S., Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005; 1(3): 183–98.
- Widmer R.J., Lerman A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014; 2014(3): 291–308. https://doi.org/10.5339/gcsp.2014.43
- Han T., Lai Y., Jiang Y., Liu X., Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading to microvascular leakage and release of pro-inflammatory cytokines. PeerJ. 2021; 9: e11892. https://doi.org/10.7717/peerj.11892
- Siragusa M., Thole J., Bibli S.I., Luck B., Loot A.E., de Silva K., et al. Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J. 2019; 38(17): e100938. https://doi.org/10.15252/embj.2018100938
- Марченко В.А., Зелинская И.А., Торопова Я.Г., Мухаметдинова Д.В., Галагудза М.М., Лиознов Д.А. и др. Длительность системных нарушений вазомоторной функции эндотелия микрососудов, вызванных вирусом гриппа А(H1N1)pdm09. Регионарное кровообращение и микроциркуляция. 2023; 22(4): 74–86. https://doi.org/10.24884/1682-6655-2023-22-4-74-86 https://elibrary.ru/mmwnsf
- Бойцов С.А. Грипп, новая коронавирусная инфекция и сердечно-сосудистые заболевания. Кардиологический вестник. 2021; 16(1): 5–9. https://doi.org/10.17116/Cardiobulletin2021160115 https://elibrary.ru/zgvxkg
- Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA. 2018; 115(23): 5839–48. https://doi.org/10.1073/pnas.1804932115
- Beckman J.S., Koppenol W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996; 271(5 Pt. 1): C1424–37. https://doi.org/10.1152/ajpcell.1996.271.5.C1424.
- Babizhayev M.A., Deyev A.I. Management of the virulent influenza virus infection by oral formulation of nonhydrolized carnosine and isopeptide of carnosine attenuating proinflammatory cytokine-induced nitric oxide production. Am. J. Ther. 2012; 19(1): e25–47. https://doi.org/10.1097/MJT.0b013e3181dcf589
- Васина Л.В., Петрищев Н.Н., Власов Т.Д. Эндотелиальная дисфункция и ее основные маркеры. Регионарное кровообращение и микроциркуляция. 2017; 16(1): 4–15. https://doi.org/10.24884/1682-6655-2017-16-1-4-15 https://elibrary.ru/yocujf
- Viemann D., Schmolke M., Lueken A., Boergeling Y., Friesenhagen J., Wittkowski H., et al. H5N1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J. Immunol. 2011; 186(1): 164–73. https://doi.org/10.4049/jimmunol.0904170
- Teijaro J.R., Walsh K.B., Cahalan S., Fremgen D.M., Roberts E., Scott F., et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011; 146(6): 980–91. https://doi.org/10.1016/j.cell.2011.08.015
- Yu J., Sun X., Goie J.Y.G., Zhang Y. Regulation of host immune responses against influenza A virus infection by Mitogen-Activated Protein Kinases (MAPKs). Microorganisms. 2020; 8(7): 1067. https://doi.org/10.3390/microorganisms8071067
- Fontijn R.D., Volger O.L., van der Pouw-Kraan T.C., Doddaballapur A., Leyen T., Baggen J.M., et al. Expression of nitric oxide-transporting aquaporin-1 is controlled by KLF2 and marks non-activated endothelium in vivo. PLoS One. 2015; 10(12): e0145777. https://doi.org/10.1371/journal.pone.0145777
- Parmar K.M., Larman H.B., Dai G., Zhang Y., Wang E.T., Moorthy S.N., et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 2006; 116(1): 49–58. https://doi.org/10.1172/jci24787
- SenBanerjee S., Lin Z., Atkins G.B., Greif D.M., Rao R.M., Kumar A., et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004; 199: 1305–15. https://doi.org/10.1084/jem.20031132
- Turpaev K.T. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochemistry (Mosc.). 2020; 85(1): 54–67. https://doi.org/10.1134/S0006297920010058
- Азаренок А.А., Еропкина Е.М., Прочуханова А.Р., Шалджян А.А., Козлова Н.М., Козелецкая К.Н. и др. Воздействие вирусов гриппа A и их поверхностных белков на метаболизм клеток эндотелия кровеносных сосудов человека. Вопросы вирусологии. 2013; 58(3): 25–7. https://elibrary.ru/pzxtur
- Hiyoshi M., Indalao I.L., Yano M., Yamane K., Takahashi E., Kido H. Influenza A virus infection of vascular endothelial cells induces GSK-3β-mediated β-catenin degradation in adherens junctions, with a resultant increase in membrane permeability. Arch. Virol. 2015; 160(1): 225–34. https://doi.org/10.1007/s00705-014-2270-5
- Betteridge K.B., Arkill K.P., Neal C.R., Harper S.J., Foster R.R., Satchell S.C., et al. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. J. Physiol. 2017; 595(15): 5015–35. https://doi.org/10.1113/JP274167
- Taghavi S., Abdullah S., Shaheen F., Mueller L., Gagen B., Duchesne J., et al. Glycocalyx degradation and the endotheliopathy of viral infection. PLoS One. 2022; 17(10): e0276232. https://doi.org/10.1371/journal.pone.0276232
- Simionescu M. Structural biochemical and functional differentiation of the vascular endothelium. In: Risau W., ed. Morphogenesis of the Endothelium. Amsterdam: Harwood Academic; 2000: 1–21.
- Armstrong S.M., Wang C., Tigdi J., Si X., Dumpit C., Charles S., et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS One. 2012; 7(10): e47323 https://doi.org/10.1371/journal.pone.0047323
- Yang Y., Schmidt E.P. The endothelial glycocalyx: an important regulator of the pulmonary vascular barrier. Tissue Barriers. 2013; 1(1): e23494. https://doi.org/10.4161/tisb.23494
- Ferro T., Neumann P., Gertzberg N., Clements R., Johnson A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000; 278(6): L1107–17. https://doi.org/10.1152/ajplung.2000.278.6.L1107.
- Kim K., Jung H., Shin I., Choi B., Kim D. Induction of interleukin-1 beta (IL-1 β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015; 87: 1104–12. https://doi.org/10.1002/jmv.24138.
- Wang S., Le T.Q., Kurihara N., Chida J., Cisse Y., Yano M., et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J. Infect. Dis. 2010; 202(7): 991–1001. https://doi.org/10.1086/656044
- Collins T., Read M.A., Neish A.S., Whitley M.Z., Thanos D., Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995; 9(10): 899-909.
- Guan X., Yang W., Sun X., Wang L., Ma B., Li H., et al. Association of influenza virus infection and inflammatory cytokines with acute myocardial infarction. Inflamm. Res. 2012; 61(6): 591–8. https://doi.org/10.1007/s00011-012-0449-3
- Singh V., Kaur R., Kumari P., Pasricha C., Singh R. ICAM-1 and VCAM-1: Gatekeepers in various inflammatory and cardiovascular disorders. Clin. Chim. Acta. 2023; 548: 117487. https://doi.org/10.1016/j.cca.2023.117487
- George S.T., Lai J., Ma J., Stacey H.D., Miller M.S., Mullarkey C.E. Neutrophils and influenza: a thin line between helpful and harmful. Vaccines (Basel). 2021; 9(6): 597. https://doi.org/10.3390/vaccines9060597
- Tang B.M., Shojaei M., Teoh S., Meyers A., Ho J., Ball T.B., et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat. Commun. 2019; 10(1): 3422. https://doi.org/10.1038/s41467-019-11249-y
- Narasaraju T., Yang E., Samy R.P., Ng H.H., Poh W.P., Liew A.A., et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011; 179(1): 199–210. https://doi.org/10.1016/j.ajpath.2011.03.013
- Saffarzadeh M., Juenemann C., Queisser M.A., Lochnit G., Barreto G., Galuska S.P., et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012; 7(2): e32366; http://doi.org/10.1371/journal.pone.0032366
- Schleef R.R., Bevilacqua M.P., Sawdey M., Gimbrone M.A. Jr., Loskutoff D.J. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J. Biol. Chem. 1988; 263(12): 5797–803.
- Marchenko V., Mukhametdinova D., Amosova I., Lioznov D., Zhilinskaya I. Influenza A(H1N1)pdm09 virus alters expression of endothelial factors in pulmonary vascular endothelium in rats. Viruses. 2022; 14(11): 2518. https://doi.org/10.3390/v14112518
- Bussolino F., Camussi G., Baglioni C. Synthesis and release of platelet-activating factor by human vascular endothelial cells treated with tumor necrosis factor or interleukin 1 alpha. J. Biol. Chem. 1988; 263(24): 11856–61.
- Счастливцев И.В., Лобастов К.В., Цаплин С.Н., Мкртычев Д.С. Современный взгляд на систему гемостаза: клеточная теория. Медицинский совет. 2019; (16): 72–7. https://doi.org/10.21518/2079-701X-2019-16-72-77 https://elibrary.ru/smgyfk
- Visseren F.L., Bouwman J.J., Bouter K.P., Diepersloot R.J., de Groot P.H., Erkelens D.W. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb. Haemost. 2000; 84(2): 319–24.
- Zelaya H., Tada A., Vizoso-Pinto M.G., Salva S., Kanmani P., Agüero G., et al. Nasal priming with immunobiotic Lactobacillus rhamnosus modulates inflammation-coagulation interactions and reduces influenza virus-associated pulmonary damage. Inflamm. Res. 2015; 64(8): 589–602. https://doi.org/10.1007/s00011-015-0837-6
- Cesari M., Pahor M., Incalzi R.A. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010; 28(5): e72–91. https://doi.org/10.1111/j.1755-5922.2010.00171.x
- Слуханчук Е.В., Бицадзе В.О., Хизроева Д.Х., Солопова А.Г., Цибизова В.И., Якубова Ф. и др. Роль тромбоцитов в противовирусном иммунитете. Акушерство, гинекология и репродукция. 2022; 16(2): 204–12. https://doi.org/10.17749/2313-7347/ob.gyn.rep.2022.305 https://elibrary.ru/twhjna
- Iba T., Levi M., Thachil J., Levy J.H. Disseminated intravascular coagulation: the past, present, and future considerations. Semin. Thromb. Hemost. 2022; 48(8): 978–87. https://doi.org/10.1055/s-0042-1756300
- Jansen A.J.G., Spaan T., Low H.Z., Di Iorio D., van den Brand J., Tieke M., et al. Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Adv. 2020; 4(13): 2967–78. https://doi.org/10.1182/bloodadvances.2020001640
- Панина И.Ю., Румянцев А.Ш., Меншутина М.А., Ачкасова В.В., Дегтерева О.А., Тугушева Ф.А. и др. Особенности функции эндотелия при хронической болезни почек. обзор литературы и собственные данные. Нефрология. 2007; 11(4): 28–46. https://elibrary.ru/jtygjh
- Kim K.S., Jung H., Shin I.K., Choi B.R., Kim D.H. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015; 87(7): 1104–12. https://doi.org/10.1002/jmv.24138
- Choreño-Parra J.A., Jiménez-Álvarez L.A., Cruz-Lagunas A., Rodríguez-Reyna T.S., Ramírez-Martínez G., Sandoval-Vega M., et al. Clinical and immunological factors that distinguish COVID-19 from pandemic influenza A(H1N1). Front. Immunol. 2021; 12: 593595. https://doi.org/10.3389/fimmu.2021.593595
- Sumikoshi M., Hashimoto K., Kawasaki Y., Sakuma H., Suzutani T., Suzuki H., et al. Human influenza virus infection and apoptosis induction in human vascular endothelial cells. J. Med. Virol. 2008; 80(6): 1072–8. https://doi.org/10.1002/jmv.21185
- Cassina A.M., Hodara R., Souza J.M., Thomson L., Castro L., Ischiropoulos H., et al. Cytochrome c nitration by peroxynitrite. J. Biol. Chem. 2000; 275(28): 21409–15. https://doi.org/10.1074/jbc.M909978199
- Halder U.C., Bagchi P., Chattopadhyay S., Dutta D., Chawla-Sarkar M. Cell death regulation during influenza A virus infection by matrix (M1) protein: a model of viral control over the cellular survival pathway. Cell Death Dis. 2011; 2(9): e197. https://doi.org/10.1038/cddis.2011.75
- Winn R.K., Harlan J.M. The role of endothelial cell apoptosis in inflammatory and immune diseases. J. Thromb. Haemost. 2005; 3(8): 1815–24. https://doi.org/10.1111/j.1538-7836.2005.01378.x
- Шевченко Ю.Л., Стойко Ю.М., Гудымович В.Г. Эндотелий как мишень патологического воздействия вирусной инфекции. Вестник Национального медико-хирургического центра им. Н.И. Пирогова. 2022; 17(2): 11–6. https://doi.org/10.25881/20728255_2022_17_2_11 https://elibrary.ru/yzfzkv
- Gui R, Chen Q. Molecular events involved in influenza A virus-induced cell death. Front. Microbiol. 2022; 12: 797789. https://doi.org/10.3389/fmicb.2021.797789
- Zhirnov O.P., Konakova T.E., Wolff T., Klenk H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002; 76(4): 1617–25. https://doi.org/10.1128/jvi.76.4.1617-1625.2002
- Stasakova J., Ferko B., Kittel C., Sereinig S., Romanova J., Katinger H., et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005; 86(Pt. 1): 185–95. https://doi.org/10.1099/vir.0.80422-0
- Wang X., Zheng T., Lin L., Zhang Y., Peng X., Yan Y., et al. Influenza A virus induces autophagy by its hemagglutinin binding to cell surface heat shock protein 90AA1. Front. Microbiol. 2020; 11: 566348. https://doi.org/10.3389/fmicb.2020.566348
- Othumpangat S., Noti J.D., McMillen C.M., Beezhold D.H. ICAM-1 regulates the survival of influenza virus in lung epithelial cells during the early stages of infection. Virology. 2016; 487: 85–94. https://doi.org/10.1016/j.virol.2015.10.005
- Tinoco R., Deiro M., Lin M., Bradley L. P-selectin regulation of T cell immunity during influenza virus infection (49.14). J. Immunol. 2011; 186(1 Suppl.): 49.14. https://doi.org/10.4049/jimmunol.186.Supp.49.14
- Short K.R., Veldhuis Kroeze E.J., Reperant L.A., Richard M., Kuiken T. Influenza virus and endothelial cells: a species specific relationship. Front. Microbiol. 2014; 5: 653. https://doi.org/10.3389/fmicb.2014.00653
- Garcia C.C., Russo R.C., Guabiraba R., Fagundes C.T., Polidoro R.B., Tavares L.P., et al. Platelet-activating factor receptor plays a role in lung injury and death caused by Influenza A in mice. PLoS Pathog. 2010; 6(11): e1001171. https://doi.org/10.1371/journal.ppat.1001171
- Morichi S., Morishita N., Takeshita M., Ishida Y., Oana S., Yamanaka G., et al. Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) levels in the cerebrospinal fluid of children with influenza-associated encephalopathy. J. Infect. Chemother. 2017; 23(2): 80–4. https://doi.org/10.1016/j.jiac.2016.10.007
- Schmitz N., Kurrer M., Bachmann M.F., Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 2005; 79(10): 6441–8. https://doi.org/10.1128/JVI.79.10.6441-6448.2005
- Bawazeer A.O., Rosli S., Harpur C.M., Docherty C.A., Mansell A., Tate M.D. Interleukin-1β exacerbates disease and is a potential therapeutic target to reduce pulmonary inflammation during severe influenza A virus infection. Immunol. Cell Biol. 2021; 99(7): 737–48. https://doi.org/10.1111/imcb.12459
- Жилинская И.Н., Марченко В.А., Харченко Е.П. Сравнение фрагментов, мимикрирующих белки системы гемостаза человека, в белках вирусов гриппа А/H1N1 и коронавирусов. Молекулярная генетика, микробиология и вирусология. 2022; 40(4): 43–6. https://doi.org/10.17116/molgen20224004143 https://elibrary.ru/mwqoig
- Гольдштейн Э.М. Смертность от болезней системы кровообращения и болезней органов дыхания, ассоциированная с гриппом, в российской федерации во время сезонов гриппа c 2013-2014 до 2018-2019. Международный журнал прикладных и фундаментальных исследований. 2019; (12-1): 9–16. https://doi.org/10.17513/mjpfi.12945 https://elibrary.ru/dhthqt
Дополнительные файлы

