



Cellular proteins as potential targets for antiretroviral therapy
- Authors: Bobkova M.R.1
-
Affiliations:
- I. Mechnikov Research Institute for Vaccines and Sera
- Issue: Vol 68, No 6 (2023)
- Pages: 488-504
- Section: REVIEWS
- URL: https://virusjour.crie.ru/jour/article/view/16597
- DOI: https://doi.org/10.36233/0507-4088-207
- EDN: https://elibrary.ru/klgwak
- ID: 16597

Cite item

Abstract
The review article conducts an in-depth analysis of information gleaned from a comprehensive literature search across Scopus, Web of Science, and MedLine databases. The focal point of this search revolves around the identification and exploration of the mechanisms orchestrated by host cell factors in the replication cycle of the human immunodeficiency virus (HIV-1, Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodeficiency virus-1). The article delves into two primary categories of proteins, namely HIV dependence factors (such as CypA, LEDGF, TSG101) and restriction factors (including SERINС5, TRIM5α, APOBEC3G), providing illustrative examples. The current understanding of the functioning mechanisms of these proteins is elucidated, and an evaluation is presented on the potential development of drugs for treating HIV infection. These drugs aim to either inhibit or stimulate the activity of host factors, offering insights into promising avenues for future research and therapeutic advancements.
Keywords
Full Text
Advances in antiretroviral therapy (ART), based on the use of drugs that directly target the enzymes of the virus, have radically changed the HIV epidemic from imminently fatal to controlled, restoring the life expectancy and quality of life for infected people. Despite this remarkable progress, ART still has problems that have yet to be solved to this day, nearly 30 years after the introduction of effective direct-acting tritherapy (triple antiretroviral therapy) regimens. The main problems are as follows: 1) the impossibility of completely curing the HIV infection due to the phenomenon of latency underlying the pathogenesis of this disease; 2) as a result of the previous point, the necessity for lifelong treatment; 3) the requirement of high adherence, difficult to fulfill for many patients; 4) progression of immune system hyperactivation and its consequences, despite the high virological efficacy of the treatment; 5) the presence of side effects even from modern drugs; 6) the formation of drug-resistant variants of the virus.
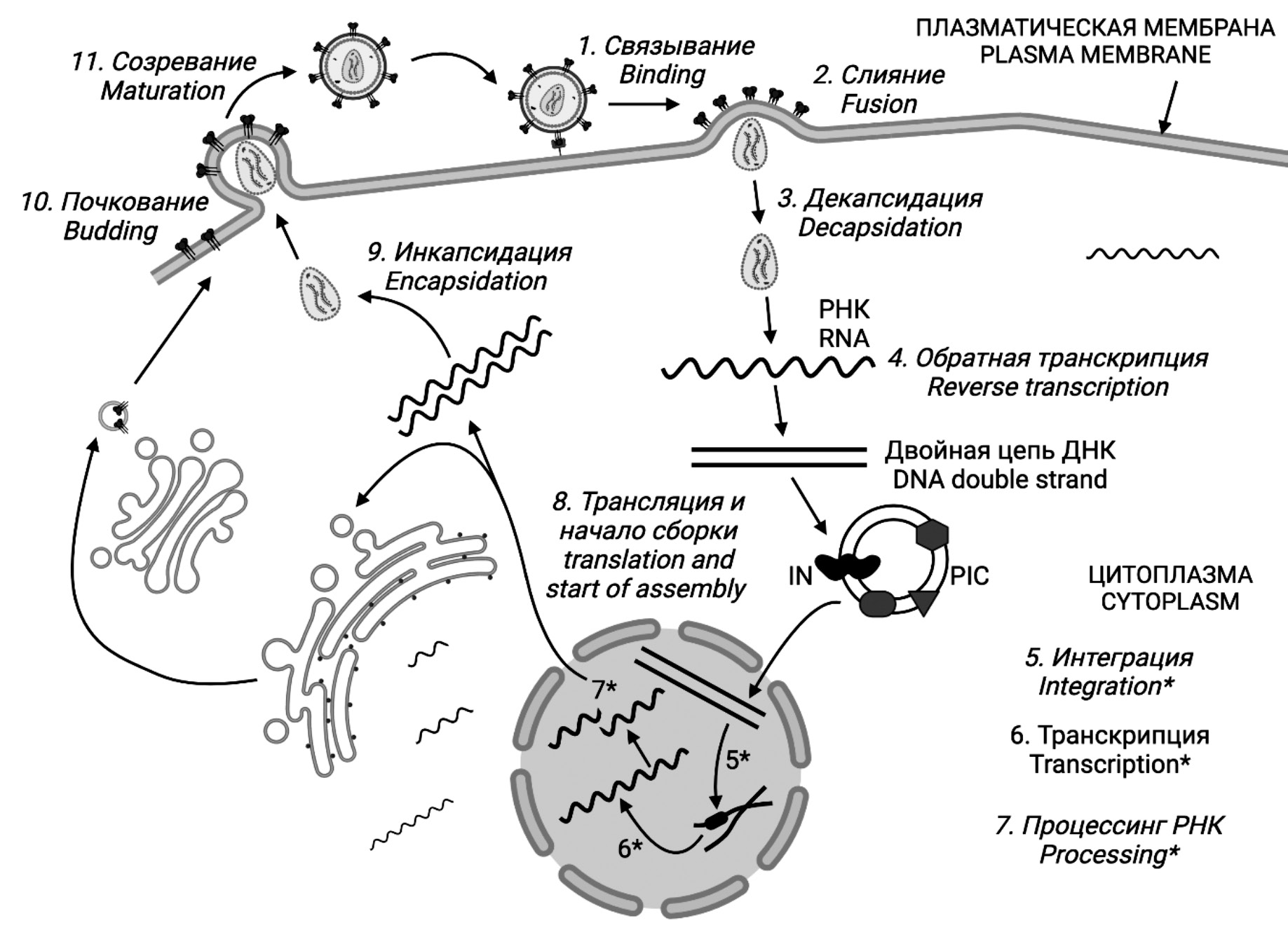
The latter phenomenon is related to the peculiarity of the HIV life cycle, which includes the reverse transcription stage (Fig. 1). A significant number of errors made by the HIV enzyme reverse transcriptase (RT) affect all regions of the genome, including the positions that determine the binding of drug molecules as ART targets. As a result, mutations occur that develop drug resistance and reduce the efficacy of ART.

Fig. 1. HIV life cycle. Figures 1–10 were created in the BioRender program (BioRender.com).
Рис. 1. Жизненный цикл ВИЧ. Рисунки 1‒10 созданы в программе BioRender (BioRender.com).
The list of ART targets is limited and includes three HIV enzymes: RT, integrase (IN) and protease (PR), as well as viral proteins involved in the infection of the cell at the stages of attachment (binding) and fusion (Fig. 1). The phenomenon of drug resistance occurs with respect to all of them, although with different frequency. Over the course of life, about 40% of patients develop ART-resistant HIV strains, which requires changing treatment regimens, usually to more expensive ones. Furthermore, resistant HIV variants can be transmitted by contact and cause infection, thus rendering the first-line ART regimen to be evidently ineffective. Innovative drugs directed against the same targets, although less often, also cause the formation of resistant viruses and generally do not cancel the phenomenon.
All of this prompts the search for fundamentally new approaches to the treatment of HIV infection, among which, in recent years, the possibility of developing drugs that would target the interaction of HIV with cellular proteins and structures important for viral reproduction has been increasingly discussed [1–7]. The intention of this approach is to create therapeutic alternatives for the treatment of viral infections that do not cause drug resistance because they target host proteins (host-targeted antivirals (HTA))that are genetically more stable than viral proteins.
Undoubtedly, host cell replicative and biosynthetic mechanisms play a critical role in the life cycle of all viruses, which are intrinsically intracellular parasites. This brief review will discuss the current state of research in this area as it relates to HIV, including the pro-viral and antiviral cellular factors that function at different stages of the viral infectious cycle and certain approaches to drug development targeting these factors.
A lot of publications on this topic started to be released in 2008 with the sensational work of Harvard scientists [8], who conducted a full-genome screening of the human genome using a library of more than 21,000 small interfering RNAs (siRNAs) in search of host factors involved in HIV reproduction. Sequential silencing of genes followed by assessment of p24 antigen production in a model system based on HeLa-derived cells yielded a staggering result. 274 human cell proteins were shown to be involved in HIV replication in one way or another, with only 37 (13%) of them known prior to the results of this experiment. This participation was realized at all stages of the HIV life cycle – from virus attachment to the cell to budding of new virions, and was manifested by positive regulation of HIV production; such factors have been given the somewhat inadequate name of HIV-dependency factors (HDFs).
Strictly speaking, the amount of detected HDFs was striking, but not the fact of their existence, as it is obvious that a virus possessing such minimal capabilities must resort to the assistance of mediator proteins for the realization of all replication stages. Subsequent studies have shown that it is not just a question of adapting the processes of virus replication in the cell to the cellular machinery, but also of active interference of HIV in a wide range of cellular processes, which include endomembrane remodeling, polymerization and organization of the cytoskeleton, modulation of gene and host protein expression, apoptosis and cell division, evasion of the immune response and much more [2, 6, 7, 9]. Not all HDFs have been studied in detail, but some of them can already be considered as potential targets for therapy.
The topic garnered more interest as the data on the existence of another group of proteins, although not so numerous but very promising from the point of view of developing therapeutic agents, accumulated. The proteins in question are the so-called restriction factors (RFs) - human proteins capable of blocking the replication of HIV in cells. The mechanisms by which they do this are extremely intricate and diverse, and are currently the subject of the most active research.
Some examples of proteins belonging to both groups of HIV enablers and detractors will be listed below in an order that roughly corresponds to the order of events of virus replication in the cell. To put it briefly, the HIV replication cycle involves the following stages: 1) attachment of viral particles followed by 2) fusion of cellular and viral membranes with the help of cell receptors; 3) decapsidation (uncoating) of viral particles; 4) reverse transcription with the help of HIV reverse transcriptase and formation of complementary DNA (cDNA); 5) formation of preintegration complex (PIC) and integration of cDNA into cell chromatin with the help of HIV integrase; 6) transcription with the help of cellular RNA polymerase; 7) RNA processing (splicing) and its export into the cytoplasm; 8) protein synthesis and assembly of new viral particles; 9) encapsidation with the formation of the internal structure of the particle; 10) budding of particles from the cell with the simultaneous attachment of envelope proteins; 11) maturation with the help of HIV protease (Fig. 1).
The virion infecting a susceptible cell always contains, in addition to structural proteins, a small number of enzymes, RT, IN and PR, as well as certain cellular proteins captured by the virion at the time of budding from the cell and required in the early stages of virus replication.
Attachment. The first cellular proteins encountered by HIV are the cellular receptors (CD4 and CCR5) required for virus attachment. The presence of the former on the cell membrane is absolutely essential for infection; the absence of the second co-receptor (CCR5) in cases of a homozygous defect – deletion of CCR5D32 – protects against infection in the vast majority of cases, but leaves a rare possibility of infection by viruses tropic to the alternative co-receptor CXCR4. This phenomenon has been described in detail many times in the literature (Fig. 2) [10-12].

Fig. 2. Stages of HIV entry into the cell.
Рис. 2. Этапы проникновения ВИЧ в клетку.
A drug aimed at inhibiting the CCR5 co-receptor already exists and is used under the name “Maraviroc” (MVC); new CCR5 and CXCR4 inhibitors are in clinical trials; CD4 inhibitors are also in development and, together with co-receptor inhibitors, form a class of HIV receptor antagonists.
In addition to the well-known surface receptors CD4 and CCR5, several additional factors have been identified whose function is critical for HIV infection of cells.
HIV is known to capable of infecting cells not only by virus-cell interaction, but also by direct cell-to-cell contact that does not require receptors. This mode of infection (cell-to-cell) is many times more efficient than the classical variant and is supported by ALCAM (activated leukocyte cell adhesion molecule) glycoproteins from the immunoglobulin superfamily, which have strong adhesive properties. ALCAMs mediate intercellular adhesion and promote efficient virus dissemination through direct cell-to-cell transmission (Fig. 2) [13, 14].
Another molecule with adhesive properties is DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin). It is not present on the surface of the main targets of HIV – CD4+ T-cells, but it is abundant on macrophages and especially dendritic cells, which are the first to encounter the virus in the periphery during infection. DC-SIGN is a C-type lectin that recognizes glycoproteins on the surface of microorganisms and, in HIV infection, acts as a nonspecific co-receptor that interacts with gp120. Subsequent internalization of the DC-SIGN-gp120 complex leads to the formation of intracellular storages of virus, and after the complex is recirculated to the cell surface, infection of CD4+ T cells in lymphoid organs after the arrival of dendritic cells in them is significantly facilitated [15].
The sulfated group within its N-terminal tyrosine is very important for the function of the CCR5 co-receptor. Two cellular proteins – SLC35B2 (solute carrier family 35 member B2) and TPST2 (tyrosylprotein sulfotransferase 2) – are involved in the sulfation of this amino acid (Fig. 2). The activated sulfate donor is PAPS (3’-phosphoadenosine 5’-phosphosulfate transporter), which is synthesized in the cytosol and is transported by SLC35B2 to the lumen of the Golgi apparatus, where the resident protein TPST2 catalyzes the sulfation of the CCR5 tyrosine. As a result [14], the absence of TPST2 and/or SLC35B2 completely prevents HIV infection of cells.
The restriction factor SERINC5 (serine incorporator 5), which is found in viral particles, opposes all the above-described cellular proteins contributing to HIV infection. There is very little understanding about its natural function; it is known that this protein has a 10-link transmembrane domain and is thus related to the CCR5 co-receptor [16]. The mechanism of its participation in HIV replication is also quite unclear; there are assumptions that it is capable of binding the gp120 protein and thereby preventing the process of membrane fusion, with gp120 lingering on the membrane surface and becoming the target of HIV antibodies (Fig. 2) [4, 17, 18].
The antagonist of SERINC5 is the viral protein Nef, which is able to remove it from the membrane composition and sequester it into the endosome with subsequent degradation [19].
Decapsidation. Until recently, it was believed that upon entry of a retrovirus into the cytosol of a host cell, immediate capsid shedding occurs, followed by RNA release and subsequent reverse transcription. Nowadays, this point of view is opposed by another one, which justly draws attention to the fact that cDNA formed during reverse transcription may become a target of molecular sensors [20, 21] sensitive to the presence of DNA in the cytosol and cause an immune reaction unnecessary for the virus. This can be avoided by protecting the resulting cDNA with a protein envelope, which is why many researchers now believe that decapsidation and reverse transcription are coupled in time, with transcription beginning inside the nucleocapsid and capsid shedding occurring as the cDNA moves toward the nucleus and ending just before PIC (see below) is imported into the nucleus [3, 22] or even already inside the nucleus (Fig. 3) [23].

Fig. 3. Dependency (CypA) and restriction (TRIM5α) factors at the HIV decapsidation stage.
Рис. 3. Факторы зависимости (CypA) и рестрикции (TRIM5α) на этапе декапсидации ВИЧ.
The central link of all interactions at this stage is the viral capsid protein (CA) p24, which constitutes the inner envelope of the virion. Representatives of both groupings of cellular proteins compete for the right to make contact with it, and the best known out of them are RF TRIM5α (tripartite motif-containing protein 5) and cyclophilin from the group of HDFs. The functioning mechanism of both of them is still insufficiently studied, and only a number of reasonable assumptions about the events involving them are presented below.
TRIM5α directly binds CA molecules of the nucleocapsid of viruses that have just entered the cytoplasm after the fusion of the viral and cell membranes is completed. Dimerization of CA disrupts the capsid structure and causes its accelerated fragmentation, thereby cancelling reverse transcription (Fig. 3) [3, 24]. TRIM5α protein is present in all primates, but its function described above is species-specific: TRIM5α from Old World monkeys, such as the rhesus macaque, restricts a wide range of retroviruses, including HIV, while human TRIM5α is unable to effectively inhibit HIV [3], although there is evidence of polymorphic variants of TRIM5α associated with the delayed progression of HIV infection [25].
Perhaps the reason for the insufficient effect of TRIM5α is the presence of efficiently working cyclophilin (CypA) in human cells. This protein from the chaperone group is present in the cell in significant amounts and different forms, taking part in the processes of protein molecule folding. It can be found at different stages of HIV replication and is always present in the composition of viral particles, apparently in order to start its activity immediately after infection of the cell. It is assumed that the increase of HIV infectivity in the presence of CypA is explained by its ability to interact with CA, thus increasing capsid stability, which, in turn, allows keeping cDNA intact until it crosses the nuclear membrane [5, 22, 23]. A simple explanation of this fact is that CypA competes with TRIM5α for binding to CA; a slightly more complex, but also quite probable, explanation is the direct TRIM5α-CypA interaction, which creates a steric hindrance to its contact with CA (Fig. 3) [26]. The intention of future ART drug development includes the creation of non-immunosuppressive CypA inhibitors as well as increasing the efficiency of TRIM5α-CypA binding [6, 26].
Cell cytoskeleton. The size of viral and subviral components of virions entering the cell does not allow them to diffuse freely in the densely populated cytoplasm, so the early stages of the HIV life cycle critically depend on the processes of cytoplasmic transport.
One of the cell proteins involved in the organization of this process is the contractile protein actin. Its microfilaments perform their function at the cell periphery and are possibly related to the process of endocytosis of viral particles (see above) [6].
The role of microtubules has been repeatedly discussed in relation to at least two aspects of the HIV life cycle, membrane fusion and intracellular trafficking of incoming capsids. The cellular proteins associated with trafficking are MAP1A and MAP1S (microtubule-associated proteins) and dynein, which make up the bulk of microtubules. The only known mechanism of their functioning is that they capable of binding ubiquitous CA as part of all viral components and directing them in the desired course, i.e. towards the cell nucleus [6, 23].
Since cytoplasmic trafficking elements constitute a significant part of host cell proteins and are essential for all intracellular processes, it is rather difficult to imagine them as therapeutic targets (e.g., by depolymerization). Given that microfilaments and microtubules are physically and functionally intertwined, the interaction between HIV and the cytoskeleton appears to be even more complex and requires further investigation.
Reverse transcription. Two RFs, APOBEC3G and SAMHD1, have gained the most prominence in this crucial stage of HIV replication.
APOBEC3G (apolipoprotein B mRNA editing enzyme, catalytic subunit 3G) functions as a deaminase during cDNA synthesis, converting deoxycytidine (dC) to deoxyuridine (dU) as part of the minus-chain. In the next stage, the resulting plus-chain DNA contains many G-A hypermutations, leading to the formation of premature stop codons and the production of aberrant viral transcripts that are eventually degraded (Fig. 4).

Fig. 4. The restriction factors APOBEC3G and SAMHD1 at the HIV reverse transcription stage.
Рис. 4. Факторы рестрикции APOBEC3G и SAMHD1 на этапе обратной транскрипции ВИЧ.
The APOBEC3G protein has its own antagonist among HIV proteins, Vif, also found within virions. By binding to APOBEC3G, Vif mediates its proteosomal degradation using the ubiquitination mechanism [4, 19, 27].
The cellular protein SAMHD1 (sterile alpha motif domain-, HD domain-containing protein 1), acting as a phosphohydrolase, converts nucleotide triphosphates (dNTPs) into deoxynucleotides, thereby depleting the resource for cDNA synthesis (Fig. 4). Furthermore, the resulting decrease in DNA levels in the cytosol protects the cell from activation of molecular sensors and subsequent unwanted interferon secretion and chronic inflammation [3, 6, 23]. HIV-1 does not have a SAMHD1 antagonist protein, but HIV-2 has the Vpx protein, which is involved in SAMHD1 degradation [3, 27].
Formation of the pre-integration complex. After the synthesis of cDNA molecules is completed, they must be incorporated into the chromosomal DNA of the cell. Unlike other retroviruses, which usually infect cells in the mitotic stage, when the nuclear membrane is absent and DNA is easily accessible, HIV has the ability to infect non-dividing cells, i.e. cells in the interphase period. Since the duration of this phase is much longer than mitosis, this capability gives the virus a tremendous advantage, providing it with a huge number of targets and the highest level of replication. In non-dividing cells, the nucleus is surrounded by a dense envelope, the nuclear membrane, so passive diffusion does not solve the issue and requires active cDNA transport. The pre-integration stage and the actual integration are the central and most complex organized events of HIV replication.
Pre-integration begins with the formation of a minimal structure, the intasome, consisting of cDNA and the IN enzyme; initially, IN has a dimeric structure, and at the stage of integration as part of a functional PIC, it takes the form of a tetramer [28]. IN molecules bring together the ends of the cDNA, after which an open ring is formed, with long terminal repeats (LTRs) at each end. After attachment of several core viral and cellular proteins, a PIC is formed (Fig. 5).

Fig. 5. Involvement of nucleoporins in the translocation of the HIV pre-integration complex.
Рис. 5. Участие нуклеопоринов в транслокации преинтеграционного комплекса ВИЧ.
Within the PIC, the first stage of HIV integration, 3’-processing of the cDNA, occurs in the cytoplasm, with IN activity playing central role. During 3’-processing, two nucleotides are removed at each of the 3’-ends of viral DNA in PIC to form free hydroxyl (OH)-groups, and the cDNA enters the nucleus in a processed form. At this stage, a problem arises that requires assistance from the cell. The problem in question is that PIC is surrounded by a noticeable number of linear cDNA molecules, which can easily self-integrate with the help of IN and thus reduce the efficiency of the subsequent true integration. This phenomenon is prevented with the help of cellular BAF (barrier-to-auto integration factor), a DNA-binding protein capable of condensing DNA [29]. Protection against such suicidal events is also provided by several DNAases associated with the endoplasmic reticulum [30].
Translocation of the pre-integration complex. After the 3’-processing is complete, the PIC begins to move towards the cell nucleus. PIC translocation is ensured by interaction of karyophilic viral proteins containing special sequences of amino acids which are nuclear localization signals (NLS) with cellular proteins functioning at all stages of nuclear transport. Viral proteins with NLS include MA, IN, and the non-structural HIV protein Vpr [31].
The nuclear membrane has pores (also known as the nuclear pore complex, NPC), mainly consisting of highly conserved nucleoporins (NUPs) [32]. They provide transport of large hydrophilic molecules with a mass of more than 40 kDa, while all other molecules penetrate into the nucleus by passive diffusion. Depending on their function, NUPs can be located either in the cytosol or in the nucleoplasm.
About 30 proteins of this group form an NPC with a ring structure: a cytoplasmic ring with 50 filaments (Nup358 is an example), a nuclear ring (nuclear basket of 8 filaments) including Nup153, and a tunnel between them (Nup170, Nup155, etc.) [32]. The internal pore size is much smaller (about 8 nm) than the PIC diameter (~28 nm) [33], so PIC has to literally squeeze through the channels in the membrane, while energy costs are provided by adenosine triphosphate (ATP).
For the process of translocation, PIC can directly contact NPC components or use soluble transporter proteins from the group of karyopherins [22]. The natural function of these proteins is to transport mRNA splicing factors into the cell nucleus; in an HIV-infected cell, the best known HIV helpers are the karyopherins TNPO3 (transportin-3) and CPSF6 (cleavage and polyadenylation specificity factor 6). Only CPSF6 appears to interact directly with CA, which does not have its own NLS, and the task of TNPO3 is to perform cleavage and polyadenylation of CPSF6. Alternatively, Nup358, which also has an affinity for CA, may act as an alternative to these proteins [34]. Finally, the same CypA is actively involved in the integration process, anchoring the PIC on the cytoplasmic side of the nuclear membrane and guiding it to the nuclear pore; the viral protein Vpr is also thought [22, 31] to be somehow involved (Fig. 5).
Passage through the pore is facilitated by several NUPs, each of which interacts with one or more PIC components. On the nuclear side of the membrane, PIC is encountered by the NUP153 protein, which localizes at the end of the NPC tunnel and directly binds IN, CA, and Vpr of HIV-1.
It is important to note that the process of PIC translocation involves a large number of participants both on the virus side (not all of them are listed here, even among the well-studied ones) and on the cell side, with the functions of each of them overlapping, and at each stage there is a choice in favor of one or another component depending on the state of the virus replication process.
All of the above factors ultimately contributing to HIV-1 replication can be categorized as HDFs. To date, only one intracellular RF that inhibits PIC translocation has been described, which is the MX2 protein (from Myxovirus resistance, aka GTPase MxB). Interferon-inducible MX2 has an affinity for CA and is concentrated on the cytoplasmic side of the nuclear membrane [35]. Little is known about its mechanisms of action; MX2 inhibits different strains of HIV-1 as well as other primate lentiviruses, but is minimally active against non-primate lentiviruses. Some strains of HIV-1 exhibit natural resistance to MX2 without any obvious effects on viral fitness [36].
True HIV integration. Once inside the nucleus, HIV-1 PICs begin a slow diffusion movement and predominantly concentrate at the periphery of the nucleus [37], not too far from the nuclear pores. The sequence of chromosomal DNA at the provirus integration site is not essential, but it would be incorrect to call the localization of this process completely random. The accessibility of DNA, i.e., the surrounding chromatin structure, seems to take priority in the choice of the integration site. In non-dividing cells, much of the cell’s DNA is in a condensed state (heterochromatin) and bound to histones, so HIV-1 preferentially binds to decondensed chromatin (euchromatin), and the hot spots for integration are transcriptionally active regions of DNA freed from histones [38]. Apparently, this not only facilitates the process of incorporation, but also gives the virus an evolutionary advantage, providing it with a high level of transcription in tandem with the target genome [6].
Interestingly, the cellular genes that are most frequently targeted for HIV-1 provirus insertion, the so-called recurrent integration genes, are also concentrated at the periphery of the nucleus and in contact with nuclear pores. Such organization of this process makes it possible to minimize the time required for cDNA integration itself.
This process is largely regulated by capsid and CA-interacting proteins [22, 39], once again indicating that cDNA translocation into the nucleus and integration are well coordinated. The selection of the integration locus can be organized hierarchically: topological localization is determined by the NPC component NUP153, whereas the cellular proteins CPSF6, LEDGF/p75 (lens epithelium-derived growth factor) and INI1 (Integrase interactor protein 1) [37, 40], as well as, of course, IN HIV-1 itself, play a dominant role in selecting the site of transcriptionally active chromatin and subsequent incorporation (Fig. 6).

Fig. 6. Integration of HIV proviral DNA.
Рис. 6. Интеграция провирусной ДНК ВИЧ.
It is believed that CPSF6, which in association with CA at the translocation stage ensured PIC penetration into the nucleus, plays a crucial role in PIC localization in the nucleus by directing it to euchromatin [40]. As for INI1, certain speculations [41] suggest that this factor may stabilize PIC in the host cell, maintaining IN in a stable conformation that prevents nonspecific interactions and self-integration. The BAF protein, which is responsible for the prevention of self-integration within PIC [29], continues to fulfill this function at the integration stage as well [28, 31, 39].
The most studied protein LEDGF/p75 serves as a link (or bridge) between the pre-integration complex and host DNA. The sequence of binding of LEDGF/p75 to host DNA and PIC remains unclear; however, regardless of the sequence of events, it is thought that the presence of LEDGF/p75 causes IN dimers to converge with each other to form a tetramer. This allosteric effect leads to the activation of IN, which then proceeds to fulfill its main function, the strand transfer reaction [29, 42]. It consists of using the previously formed hydroxyl groups of IN to cleave chromosomal DNA at the selected site and simultaneously connect its 5’-phosphate groups to the ends of viral cDNA. The intermediate result is a DNA region that has breaks and free 5’-ends of viral DNA; this temporary defect is repaired by host cell functions to form an integrated provirus [29, 42].
LEDGF/p75 began to garner interest more than a decade ago [43] and was used in the development of a group of drugs that target the interface between the active center of IN and the LEDGF binding domain (LEDGINs). The action of these inhibitors is based on an allosteric effect and manifests itself in two phenomena: early, i.e., an obvious decrease in IN activity and provirus integration, and late, which leads to morphological changes in the produced viral particles and disturbances in the virus core structure due to an increase in IN multimerization [43–46], with the ribonucleoprotein being outside the core. Such disrupted particles account for up to 70% and are unable to ensure the normal course of virus propagation events during the subsequent round of infection [46]. In preclinical trials, the negative effect of certain LEDGINs with respect to viral reservoir formation has also been confirmed [47], and this brings up the possibility of using these drugs for the functional cure of HIV infection [47, 48] already in the near future.
Transcription. HIV does not have its own capabilities for transcription of proviral DNA, and for transcription of its genome, the virus attracts all the necessary components of the cellular apparatus, primarily RNA polymerase. In order to recruit them in favor of working for the virus, the most important of the HIV regulatory factors, the Tat protein, is used. Its activity determines the level of HIV transcription, and the absence of Tat leads proviral DNA into a state of latency. The events that occur during this process have been repeatedly described in other literature [49-52], therefore it is unnecessary to further discuss them here.
In short, the signal for the initiation of RNA transcribing is the contact between the activator proteins SP1 (specificity protein 1) and NF-κB (nuclear factor [kappa]B), each of which is located in the corresponding region of the LTR promoter. Following that, the RNA polymerase (RNA-PII), having recognized the TATA-box, starts RNA synthesis, but in the absence of Tat, the synthesis is brought to an abrupt halt, and the result of this transcription stage is a short RNA fragment forming a TAR loop (Fig. 7).

Fig. 7. Activity of host factors at the HIV transcription stage.
Рис. 7. Активность хозяйских факторов на этапе транскрипции ВИЧ.
With the presence of Tat, the situation changes drastically, as it triggers the formation of the P-TEFb (positive transcription elongation factor b) complex, including CDK9 (cyclin-dependent kinase 9) and CycT (Cyclin T). The interaction of TAR/Tat/P-TEFb with apathetic RNA-PII causes a number of its modifications (mainly phosphorylation) and makes it transition to its active state, after which the polymerase proceeds to undergo productive elongation with the formation of full-length HIV mRNA.
All of the above (as well as unspecified) cellular transcription factors contribute to HIV mRNA formation and therefore, are HDFs by definition. The fate of the resulting mRNAs, however, is not always favorable, as RFs capable of leading to RNA degradation have been found at the transcription stage.
One of those RFs is RNase-L, a mediator of antiviral activity induced by interferon-1. This enzyme has a wide range of activities related to the regulation of ribosomal and viral RNAs and, in particular, is capable of exhibiting ribonuclease properties [53]. The efficient degradation of HIV mRNA with the participation of RNase-L may prevent HIV from entering the productive reproduction cycle and contribute to the maintenance of its latent state.
Another factor that causes degradation of HIV mRNA is ZAP (zinc finger antiviral protein). This protein has a broad antiviral activity and restricts not only retroviruses but also a multitude of other RNA and DNA viruses. The mechanism of its functioning remained unclear for a long time, and it only recently became apparent that the target of ZAP is CpG dinucleotides in viral RNA, the recognition of which is followed by the recruitment of components of the exosomal complex for RNA degradation (Fig. 7) [53].
Another restriction mechanism utilizes the Kruppel-associated box (KRAB)-interacting protein 1 (KAP1), which weakens the interaction of RNA-PII with the P-TEFb complex and thus retains it within the promoter, reducing the efficiency of elongation [54].
Nuclear export. Upon completion of transcription, only one type of RNA molecules is formed, with their size corresponding to those of the provirus. As it turned out, the fate of newly synthesized full-size RNA molecules is not predetermined, and the same molecules can act as matrix or genomic RNA, being incorporated into future viral particles during assembly [2, 51]. The existence of such unspliced HIV-1 transcripts contradicts the internal organization of the cell, so a special strategy involving the Rev viral protein and cellular factors was developed to remove them.
Upon completion of transcription, the full-length HIV-1 mRNA transcript first undergoes splicing to form HIV-1 Rev, Tat and Nef mRNA proteins [51], after which Rev mRNA is transported to the cytoplasm for translation of Rev. Rev protein, which has NLS, returns to the nucleus with the help of importins. In the late stages of HIV-1 reproduction, when a sufficient concentration of Rev is reached, it begins to fulfill its main function of removing unspliced mRNAs from the nucleus.
Rev contains a nucleus export signal (NES), by means of which it binds a protein from the group of karyopherins CRM1 (chromosome maintenance region 1), also known as exportin-1, and releases full-length transcripts into the cytoplasm, where they become the matrix for translation of structural proteins Gag and Pol, and also become part of newly formed virions. Other splicing products are required for the formation of auxiliary proteins Vif and Vpr, as well as envelope proteins Env [23]. The same ribonucleic acid complexes (spliceosomes) used by the cell to perform this task for its own proteins are used for splicing HIV RNA, and there is evidence that the Rev protein involves cellular RNA helicases in this event [55], which act as molecular shuttles for short HIV transcripts (Fig. 8).

Fig. 8. Splicing and nuclear export of HIV mRNAs.
Рис. 8. Сплайсинг и ядерный экспорт мРНК ВИЧ.
Nuclear export of mRNA, like PIC import, requires passage through nuclear pores (NPCs) and is mediated by soluble shuttle receptors that travel between the nucleus and cytoplasm. In addition to CRM1 described above, the TAP-p15 protein [32] functions in the export stage, as well as about a dozen less studied cellular factors that also attach to mRNA to form mRNP (messenger ribonucleoprotein). Further events unfold as follows: mRNP interacts with NUPs, initiating export. After successful docking, mRNP moves along the internal channel of NPC, making contact with NUPs, and releases mRNA with the participation of cytoplasmic filaments of NPC NUP214 [32].
Virion translation and assembly. At this stage of replication, much like in the previous one, HIV is completely dependent on the cellular biosynthesis apparatus; however, using a variety of strategies, it effectively alters the ratio of viral and cellular protein production in its favor. Among the capabilities that HIV uses to increase and regulate the synthesis of its own proteins are the use of translation enhancers – an additional ribosome entry site IRES (internal ribosome entry site) and PCE (post-transcriptional control element) that enhances binding to the polysome, a leaky scanning mechanism to read two proteins with a common reading frame, ribosome scanning and so on. The most famous of HIV “inventions” is the phenomenon of stop codon “ignoring” and frameshift at the level of translation, which allows to regulate the ratio of production of structural proteins and enzymes encoded by the same pol gene.
These mechanisms, being of great interest, are not directly related to the topic of this review and are covered in detail in a number of articles [2, 19, 23, 51, 56], and here examples will be given of less known cases of HIV interaction with host factors at the stage of translation and subsequent assembly.
Once the splicing process is complete and all RNAs have been exported to the cytoplasm, the translation initiation stage occurs, which determines the rate of HIV replication in general. Initiation elements are present in each of the HIV transcripts, allowing them to be translated independently of each other and in itself enhancing the level of virus production. The Gag, Pol, Tat, and Rev proteins are synthesized separately from the highly modified Env glycoproteins, encountering them at the budding stage (Fig. 9).

Fig. 9. Host cell factors at the HIV translation stage.
Рис. 9. Факторы хозяйской клетки на этапе трансляции ВИЧ.
The efficiency of initiation is determined by the frequency of mRNA making contact with the polyribosome (polysome) and the strength of binding to it. These processes are influenced by several proteins at once. Certain proteins are able to enhance them, for example, splicing regulator 9G8, protein Sam68 (Src-associated in mitosis 68), RNA helicase A (RHA), and others, on the contrary, play the role of restriction factors, inhibiting or canceling initiation events. These include the IFITM (interferon-induced transmembrane protein) protein, which can eliminate mRNA interaction with the polysome [23]; its viral antagonist is the Nef protein. Another example is hnRNP E1 (heterogeneous nuclear ribonucleoprotein E1), which has the ability to dissociate the large and small subunits of the ribosome [2].
As the final stage of reproduction, the assembly and budding of virions, approaches, the rivalry between the two groups of proteins becomes increasingly pronounced. After reaching a sufficient concentration of biosynthesis products which are viral proteins, the process of virion assembly begins, and the main role in the organization of the internal structure of particles, RNA incorporation and promotion to the cell membrane belongs to viral proteins of the Gag group [56]. At the initial stage of assembly, viral particles are immature, and only after cleavage of the Gag-Pol precursor are all internal proteins formed, in particular CA, which forms the capsid part of the conical shaped virion.
As it was found out [57], at subsequent stages HIV actively uses for budding a complex of cellular proteins of the ESCRT (endosomal sorting complexes required for transport) apparatus involved in membrane remodeling processes. The protein CA, known for its vigorous activity, interacting with the ESCRT component – protein TSG101 (tumor susceptibility gene 101), promotes the movement of forming viral particles into endosomes with the subsequent formation of multivesicular bodies (MVB) (Fig. 10). In this form, future virions can persist in the cytoplasm until the time of budding.

Fig. 10. Assembly and budding of HIV viral particles.
Рис. 10. Сборка и почкование вирусных частиц ВИЧ.
The Env protein is synthesized as a precursor of gp160 in the endoplasmic reticulum (EPR) and undergoes maturation/glycosylation to form gp120 and gp41 with the participation of the Golgi apparatus. Its movement from the EPR to the site of final formation is assisted by the cellular Rab11-FIP1C protein (FIP1C) (Rab coupling protein) [56]. The preferred sites for the subsequent anchoring of Env in the membrane are lipid rafts enriched with cholesterol and saturated fatty acids.
The culmination occurs at the stage of budding, when the effect of several restriction factors is manifested at once. Two of them, GBP5 (guanylate binding protein 5) and MARCH8 (E3 ubiquitin ligase membrane-associated RING-CH 8), prevent the cleavage of gp160 and thereby reduce the efficiency of Env proteins incorporation into new virions; the result is the formation of noninfectious viral particles. Furthermore, MARCH8 has the ability to retain Env in the cytoplasm by a mechanism that hasn’t been fully investigated. The PSGL-1 (P-selectin glycoprotein ligand 1) protein restricts actin dynamics near the cell membrane and traps Env within it [56].
The most studied of the restriction factors at the assembly stage is tetherin (tetherin, BST-2, bone marrow stromal antigen 2). This protein has an unusual structure that includes a T-terminal transmembrane end and a C-terminal glycosylated end, which allows it to be retained in the membrane while simultaneously capturing newly formed virions. BST-2 binds lipid rafts and actin molecules together, thereby directly restricting the mobility of virions at the time when they are ready to detach from the membrane [3, 16, 19, 56] (Figure 10). Teterin significantly reduces virus-cell infection and to a lesser extent cell-to-cell infection [16]; this is the basis for the view that virus-cell infection predominantly occurs during virus transmission, while the cell-to-cell pathway predominates at the viral dissemination stage in the body.
The role of BST-2 antagonist is assumed by the Vpu viral protein, which binds the N-terminal region of the cytoplasmic domain of BST-2, removes it from the membrane and stimulates its degradation via the proteasomal or lysosomal pathway; alternatively, BST-2 is simply displaced from the virion assembly site [58]. It is interesting to note that HIV-2 lacks the Vpu protein and its function is taken over by the Nef protein [59].
This previous example is suggestive of the co-evolution of HIV and its natural hosts over tens of thousands of years. Restriction factors are cellular proteins, each of which has a function related to cell life support. The fulfillment of this function often overlaps with the replicative cycle of the virus and causes powerful inhibitory activity. This circumstance set the virus the task of adaptation, with which it ingeniously coped, in the course of evolution acquiring additional proteins, moreover, possessing the ability to overlap and support duties. Understanding the structure and function of these accessory proteins and the mechanisms of their interaction with ligands in the host cell may be important for the development of new therapies for HIV infection.
Another aspect concerning future developments involves a detailed study of the natural functions of the cellular factors themselves, both HDFs and RFs. The former should presumably be targeted for inhibition, while the latter should be stimulated to limit HIV replication.
The development of drugs with inhibitory properties is a traditional task of pharmacology, but inhibition of cellular proteins with diverse functions potentially always carries the risk of disruption of cell metabolism and subsequent toxicity, so it is obvious that of the many HDFs, a significant proportion will be “rejected” in trials, and this will take a long time. The same is true for RFs, with stimulant development having fewer examples and will require considerable creativity.
In this regard, the position of the authors proposing to test the antiviral activity of drugs used to treat other diseases, both infectious and non-infectious, is interesting. Examples of this kind exist in the history of antiretroviral drug development. For example, the legendary azidothymidine was invented as a drug with a broad antibacterial and antiparasitic effect [60]. There are reports that drugs to correct lipid metabolism (statins) are effective in preventing viral load increase in HIV-infected patients [61], although the mechanism of this effect remains unclear. The obvious advantage of non-modern drugs is that their introduction does not require many years of preliminary clinical trials.
Finally, a promising idea is that the target of antiretroviral therapy would not be the viral and cellular proteins themselves, but the possibility of their contact, the so-called virus-host interactome [7], and therefore it would ensure the interruption of key host-virus interactions. The allosteric inhibitors of LEDGINs described above, which play the role of a spacer between the viral protein IN and the cellular factor LEDGF, could possibly be an example of such an approach.
Thus, the involvement of various cellular entities in the pathogenesis of HIV infection is becoming increasingly clear. This review attempts to summarize some of the intracellular and molecular interactions of viral and cellular proteins known to date. The identification and study of the multitude of cellular proteins exploited by the virus at all stages of the life cycle presents great opportunities for ART. Cellular targets are particularly of interest because they are virtually immune to mutational changes, and thus the problem of drug resistance is significantly less relevant for them than for viral targets. Nevertheless, many questions must be answered before the available knowledge becomes the basis for the development of new drugs. It should be clarified to what extent the phenomena and processes observed so far mainly in vitro correspond to the reality in vivo; which of the activities of cellular proteins can be inhibited without noticeable damage to the metabolism of the cell itself; the function of which factors could be disabled without appreciable physiological consequences. The results of these studies shall be observed with interest in the coming years.
About the authors
Marina R. Bobkova
I. Mechnikov Research Institute for Vaccines and Sera
Author for correspondence.
Email: mrbobkova@mail.ru
ORCID iD: 0000-0001-5481-8957
Dr. Sci. (Biol.), Chief Researcher of the laboratory of biology of lentiviruses I. Mechnikov Research Institute for Vaccines and Sera, Moscow, Russia
Россия, 105064, MoscowReferences
- Roa-Linares V.C., Escudero-Florez M., Vicente-Manzanares M., Gallego-Gomez J.C. Host cell targets for unconventional antivirals against RNA viruses. Viruses. 2023; 15(3): 776. https://doi.org/10.3390/v15030776
- Bolinger C., Boris-Lawrie K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology. 2009; 6: 8. https://doi.org/10.1186/1742-4690-6-8
- Colomer-Lluch M., Ruiz A., Moris A., Prado J.G. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front. Immunol. 2018; 9: 2876. https://doi.org/10.3389/fimmu.2018.02876
- Ghimire D., Rai M., Gaur R. Novel host restriction factors implicated in HIV-1 replication. J. Gen. Virol. 2018; 99(4): 435–46. https://doi.org/10.1099/jgv.0.001026
- Puhl A.C., Garzino Demo A., Makarov V.A., Ekins S. New targets for HIV drug discovery. Drug Discov. Today. 2019; 24(5): 1139–47. https://doi.org/10.1016/j.drudis.2019.03.013
- Schaller T., Herold N. The early bird catches the worm – can evolution teach us lessons in fighting HIV? Curr. HIV Res. 2016; 14(3): 183–210. https://doi.org/10.2174/1570162x14999160224094914
- Shukla E., Chauhan R. Host-HIV-1 interactome: a quest for novel therapeutic intervention. Cells. 2019; 8(10): 1155. https://doi.org/10.3390/cells8101155
- Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008; 319(5865): 921–6. https://doi.org/10.1126/science.1152725
- Goffinet C. Cellular antiviral factors that target particle infectivity of HIV-1. Curr. HIV Res. 2016; 14(3): 211–6. https://doi.org/10.2174/1570162x14666151216145521
- Alvarez V., Lopez-Larrea C., Coto E. Mutational analysis of the CCR5 and CXCR4 genes (HIV-1 co-receptors) in resistance to HIV-1 infection and AIDS development among intravenous drug users. Hum. Genet. 1998; 102(4): 483–6. https://doi.org/10.1007/s004390050726
- Cohn S.K. Jr., Weaver L.T. The black death and AIDS: CCR5-Delta32 in genetics and history. QJM. 2006; 99(8): 497–503. https://doi.org/10.1093/qjmed/hcl076
- Dean M., Carrington M., Winkler C., Huttley G.A., Smith M.W., Allikmets R., et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996; 273(5283): 1856–62. https://doi.org/10.1126/science.273.5283.1856
- Tsui C.K., Gupta A., Bassik M.C. Finding host targets for HIV therapy. Nat. Genet. 2017; 49(2): 175–6. https://doi.org/10.1038/ng.3777
- Park R.J., Wang T., Koundakjian D., Hultquist J.F., Lamothe-Molina P., Monel B., et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Gen. 2017; 49(2): 193–203. https://doi.org/10.1038/ng.3741
- Blumenthal R., Durell S., Viard M. HIV entry and envelope glycoprotein-mediated fusion. J. Biol. Chem. 2012; 287(49): 40841–9. https://doi.org/10.1074/jbc.r112.406272
- Hadpech S., Moonmuang S., Chupradit K., Yasamut U., Tayapiwatana C. Updating on roles of HIV intrinsic factors: A review of their antiviral mechanisms and emerging functions. Intervirology. 2022; 65(2): 67–79. https://doi.org/10.1159/000519241
- Gonzalez-Enriquez G.V., Escoto-Delgadillo M., Vazquez-Valls E., Torres-Mendoza B.M. SERINC as a Restriction Factor to Inhibit Viral Infectivity and the Interaction with HIV. J. Immunol. Res. 2017; 2017: 1548905. https://doi.org/10.1155/2017/1548905
- Tedbury P.R., Sarafianos S.G. Exposing HIV’s weaknesses. J. Biol. Chem. 2017; 292(14): 6027–8. https://doi.org/10.1074/jbc.h117.777714
- Lopez Hernandez M., Lopez De Lucio S. Accessory regulatory proteins of HIV-1 and host restriction factors interactions. Biomed. J. Sci. Tech. Res. 2020; 31(4): 24308–12. https://doi.org/10.26717/BJSTR.2020.31.005120
- Huérfano S., Šroller V., Bruštíková K., Horníková L., Forstová J. The interplay between viruses and host DNA sensors. Viruses. 2022; 14(4): 666. https://doi.org/10.3390/v14040666
- Yin X., Langer S., Zhang Z., Herbert K.M., Yoh S., König R., et al. Sensor sensibility-HIV-1 and the innate immune response. Cells. 2020; 9(1): 254. https://doi.org/10.3390/cells9010254
- Yamashita M., Engelman A.N. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017; 25(9): 741–55. https://doi.org/10.1016/j.tim.2017.04.004
- Ramdas P., Sahu A.K., Mishra T., Bhardwaj V., Chande A. From entry to egress: strategic exploitation of the cellular processes by HIV-1. Front. Microbiol. 2020; 11: 559792. https://doi.org/10.3389/fmicb.2020.559792
- Malim M.H., Bieniasz P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012; 2(5): a006940. https://doi.org/10.1101/cshperspect.a006940
- van Manen D., Rits M.A., Beugeling C., van Dort K., Schuitemaker H., Kootstra N.A. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008; 4(2): e18. https://doi.org/10.1371/journal.ppat.0040018
- Kim K., Dauphin A., Komurlu S., McCauley S.M., Yurkovetskiy L., Carbone C., et al. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 2019; 4(12): 2044–51. https://doi.org/10.1038/s41564-019-0592-5
- Harris R.S., Hultquist J.F., Evans D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012; 287(49): 40875–83. https://doi.org/10.1074/jbc.r112.416925
- Engelman A., Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008; 4(3): e1000046. https://doi.org/10.1371/journal.ppat.1000046
- Renzi G., Carta F., Supuran C.T. The integrase: an overview of a key player enzyme in the antiviral scenario. Int. J. Mol. Sci. 2023; 24(15): 12187. https://doi.org/10.3390/ijms241512187
- Lee M.S., Craigie R. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl Acad. Sci. USA. 1998; 95(4): 1528–33. https://doi.org/10.1073/pnas.95.4.1528
- Bin Hamid F., Kim J., Shin C.G. Cellular and viral determinants of retroviral nuclear entry. Can. J. Microbiol. 2016; 62(1): 1–15. https://doi.org/10.1139/cjm-2015-0350
- Tingey M., Li Y., Yu W., Young A., Yang W. Spelling out the roles of individual nucleoporins in nuclear export of mRNA. Nucleus. 2022; 13(1): 170–93. https://doi.org/10.1080/19491034.2022.2076965
- Endsley M.A., Somasunderam A.D., Li G., Oezguen N., Thiviyanathan V., Murray J.L., et al. Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology. 2014; 454-455: 60–6. https://doi.org/10.1016/j.virol.2014.02.006
- Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010; 7(3): 221–33. https://doi.org/10.1016/j.chom.2010.02.007
- Kane M., Yadav S.S., Bitzegeio J., Kutluay S.B., Zang T., Wilson S.J., et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013; 502(7472): 563–6. https://doi.org/10.1038/nature12653
- Wei W., Guo H., Ma M., Markham R., Yu X.F. Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine. 2016; 8: 230–6. https://doi.org/10.1016/j.ebiom.2016.04.020
- Lelek M., Casartelli N., Pellin D., et al. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015; 6: 6483. https://doi.org/10.1038/ncomms7483
- Demeulemeester J., De Rijck J., Gijsbers R., Debyser Z. Retroviral integration: Site matters: Mechanisms and consequences of retroviral integration site selection. Bioessays. 2015; 37(11): 1202–14. https://doi.org/10.1002/bies.201500051
- Bedwell G.J., Engelman A.N. Factors that mold the nuclear landscape of HIV-1 integration. Nucleic Acids Res. 2021; 49(2): 621–35. https://doi.org/10.1093/nar/gkaa1207
- Saito A., Henning M.S., Serrao E., Dubose B.N., Teng S., Huang J., et al. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J. Virol. 2016; 90(15): 6918–35. https://doi.org/10.1128/jvi.00019-16
- Maillot B., Lévy N., Eiler S., Crucifix C., Granger F., Richert L., et al. Structural and functional role of INI1 and LEDGF in the HIV-1 preintegration complex. PloS One. 2013; 8(4): e60734. https://doi.org/10.1371/journal.pone.0060734
- Lapaillerie D., Lelandais B., Mauro E., Lagadec F., Tumiotto C., Miskey C., et al. Modulation of the intrinsic chromatin binding property of HIV-1 integrase by LEDGF/p75. Nucleic Acids Res. 2021; 49(19): 11241–56. https://doi.org/10.1093/nar/gkab886
- Christ F., Voet A., Marchand A., Nicolet S., Desimmie B.A., Marchand D., et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010; 6(6): 442–8. https://doi.org/10.1038/nchembio.370
- Desimmie B.A., Schrijvers R., Demeulemeester J., Borrenberghs D., Weydert C., Thys W., et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology. 2013; 10: 57. https://doi.org/10.1186/1742-4690-10-57
- Le Rouzic E., Bonnard D., Chasset S., Bruneau J.M., Chevreuil F., Le Strat F., et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology. 2013; 10: 144. https://doi.org/10.1186/1742-4690-10-144
- Vranckx L.S., Demeulemeester J., Saleh S., Boll A., Vansant G., Schrijvers R., et al. LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine. 2016; 8: 248–64. https://doi.org/10.1016/j.ebiom.2016.04.039
- Bruggemans A., Vansant G., Balakrishnan M., Mitchell M.L., Cai R., Christ F., et al. GS-9822, a preclinical LEDGIN candidate, displays a block-and-lock phenotype in cell culture. Antimicrob. Agents Chemother. 2023; 65(5): e02328-20. https://doi.org/10.1128/aac.02328-20
- Debyser Z., Bruggemans A., Van Belle S., Janssens J., Christ F. LEDGINs, inhibitors of the interaction between HIV-1 integrase and LEDGF/p75, are potent antivirals with a potential to cure HIV infection. Adv. Exp. Med. Biol. 2021;1322: 97–114. https://doi.org/10.1007/978-981-16-0267-2_4
- Romani B., Allahbakhshi E. Underlying mechanisms of HIV-1 latency. Virus Genes. 2017; 53(3): 329–39. https://doi.org/10.1007/s11262-017-1443-1
- Wang S., Qiu L., Yan X., Jin W., Wang Y., Chen L., et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology. 2012; 55(3): 730–41. https://doi.org/10.1002/hep.24809
- Bobkova M.R. HIV Latency [Latentnost’ VICh]. Moscow: Chelovek; 2021. (in Russian)
- Kuznetsova A.I., Gromov K.B., Kireev D.E., Shlykova A.V., Lopatukhin A.E., Kazennova E.V., et al. Analysis of Tat protein characteristics in human immunodeficiency virus type 1 sub-subtype A6 (Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodeficiency virus-1). Voprosy virusologii. 2022; 66(6): 452–64. https://doi.org/10.36233/0507-4088-83
- Nchioua R., Bosso M., Kmiec D., Kirchhoff F. Cellular factors targeting HIV-1 transcription and viral RNA transcripts. Viruses. 2020; 12(5): 495. https://doi.org/10.3390/v12050495
- Mousseau G., Valente S.T. Role of host factors on the regulation of tat-mediated HIV-1 transcription. Curr. Pharm. Des. 2017; 23(28): 4079–90. https://doi.org/10.2174/1381612823666170622104355
- Naji S., Ambrus G., Cimermančič P., Reyes J.R., Johnson J.R., Filbrandt R., et al. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol. Cell. Proteomics. 2012; 11(4): M111.015313. https://doi.org/10.1074/mcp.m111.015313
- Lerner G., Weaver N., Anokhin B., Spearman P. Advances in HIV-1 assembly. Viruses. 2022; 14(3): 478. https://doi.org/10.3390/v14030478
- Rose K.M. When in need of an ESCRT: The nature of virus assembly sites suggests mechanistic parallels between nuclear virus egress and retroviral budding. Viruses. 2021; 13(6): 1138. https://doi.org/10.3390/v13061138
- Sauter D. Counteraction of the multifunctional restriction factor tetherin. Front. Microbiol. 2014; 5: 163. https://doi.org/10.3389/fmicb.2014.00163
- McNatt M.W., Zang T., Bieniasz P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013; 9(4): e1003299. https://doi.org/10.1371/journal.ppat.1003299
- Pattishal K. Discovery and development of Zidovudine as the cornerstone of therapy to control human immunodeficiency virus infection. In: Adams J., Merluzzi V.J., eds. The Search for Antiviral Drugs: Case Histories from Concept to Clinic. Boston, MA: Birkhäuser; 1993.
- Drechsler H., Ayers C., Cutrell J., Maalouf N., Tebas P., Bedimo R. Current use of statins reduces risk of HIV rebound on suppressive HAART. PLoS One. 2017; 12(3): e0172175. https://doi.org/10.1371/journal.pone.0172175
Supplementary files

