



Сравнительный анализ полногеномных последовательностей изолятов вируса африканской чумы свиней (Asfarviridae: Asfivirus), выделенных на территории левобережья Днепра в 2023 году
- Авторы: Чернышев Р.С.1, Иголкин А.С.1, Зиняков Н.Г.1, Чвала И.А.1
-
Учреждения:
- ФГБУ «Федеральный центр охраны здоровья животных»
- Выпуск: Том 69, № 5 (2024)
- Страницы: 481-494
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://virusjour.crie.ru/jour/article/view/16676
- DOI: https://doi.org/10.36233/0507-4088-263
- EDN: https://elibrary.ru/tsiuzd
- ID: 16676

Цитировать

Аннотация
Введение. Отсутствие данных о полногеномных последовательностях возбудителя африканской чумы свиней (АЧС), циркулирующего на территории левобережья Днепра, ограничивает понимание динамики молекулярной эволюции вируса и характера развития текущего эпизоотического процесса в центральной России и Украине. Определение степени генетической дивергентности и филогенетического родства вируса АЧС во многом может скорректировать стратегию общей и специфической профилактики болезни.
Цель работы – поиск и описание уникальных точечных мутаций (делеций/инсерций/замен) у изолятов, выделенных от домашних свиней на территории Донецкого, Луганского и Запорожского регионов в 2023 г.; установление родства и уровня гомологии с референтными штаммами вируса АЧС генотипа II; субгенотипирование на основе маркерных областей генома.
Материалы и методы. В качестве образцов использовали культуральную суспензию костного мозга свиньи, содержащую вирус АЧС. Подготовку геномной ДНК выполняли методом очистки и концентрирования вируса с последующей экстракцией тотальной нуклеиновой кислоты фенол-хлороформным методом. Процесс высокопроизводительного секвенирования осуществляли с помощью технологии MGI. Сборку консенсусных последовательностей проводили методом картирования прочтений на референс-геном штамма Georgia 2007/1.
Результаты. Все изоляты отнесены к генотипу II, имеют монофилетическое происхождение, филогенетически относятся к кластерам «Европа» (4/5) и «Брянск 2021» (1/5), а также являются дивергентными от исходных родительских генетических вариантов, составляющих укрупненные клады. Кроме того, обнаружены многочисленные замены в локусах мультигенного семейства MGF 110, 505 и 360, кодирующих факторы вирулентности.
Заключение. На примере изучения филогении показана достоверная для дифференциации вариабельность вируса АЧС генотипа II, происходящего от референс-штамма Georgia 2007/1. Представленные данные обладают теоретической и практической значимостью при усовершенствовании национального и международного надзора за АЧС.
Полный текст
Введение
В XXI веке за относительно короткий период времени африканская чума свиней (АЧС) стала мировой проблемой свиноводства. После интродукции вируса АЧС генотипа II из Африки в Грузию (2007 г.) болезнь была зарегистрирована на территории 46 государств Европы и Азии (2007–2024 гг.), Карибского бассейна (Гаити и Доминиканская Республика, 2021 г.), принимая панзоотический характер и нанося серьезный экономический ущерб свиноводческой отрасли и охотничьему сектору неблагополучных стран и регионов [1]1. Стремительное трансграничное распространение АЧС поспособствовало развитию методологии расследования вспышек, альтернативной эпизоотологическому обследованию.
Развитие методов молекулярной биологии (секвенирование) и биоинформатики (филогенетический и эволюционный анализы) значительно повлияло на формирование нового прикладного направления – молекулярной эпизоотологии, изучающего закономерности возникновения и циркуляции генетических вариантов и групп возбудителей инфекционных болезней животных [2]. В этом отношении пространственно-временная филогеномика является эффективным инструментом внутреннего и международного надзора за АЧС.
На евроазиатском континенте наблюдался медленный темп (1,14 × 10−5 замен/сайт/год) мутаций вируса АЧС, ограничивающий проведение кластеризации [3]. Так, отмечена генетическая однородность изолятов, выделенных в 2007–2011 гг. на территории Северо-Кавказского, Южного и Северо-Западного федеральных округов Российской Федерации, при исследовании маркерных фрагментов, ранее предложенных для характеристики генотипа I [4]. В связи с этим наиболее предпочтителен полногеномный анализ с высокой разрешающей способностью для обнаружения уникальных единичных изменений и установления филогенетического родства [5]. В работах А. Mazloum и соавт. (2021 г.) продемонстрирована дивергенция между вариантами, циркулирующими в Центральной России и на Кавказе, Восточной Европе и Дальнем Востоке [6].
В результате распространения болезни значительную генетическую сегрегированность друг от друга показали три группы: вирус АЧС, циркулирующий в начале эпизоотии (Грузия, Армения, Азербайджан, Россия до 2019 г., Польша и Литва до 2015 г.); изоляты из стран Балтии, восточной Европы и Германии (Румыния, Польша и Литва с 2016 г., Латвия, Эстония, Киевская область Украины и Калининградская область России); вирус АЧС, выделенный на территории Бельгии, Венгрии, Чехии и Молдовы и восточных стран (Китай, Вьетнам, Республика Корея, Индия, Восточный Тимор, а также Дальневосточный федеральный округ России) [7, 8]. Близкое филогенетическое родство вируса, энзоотичного для европейских и азиатских стран, объясняют предположительным его заносом в Китай (2018 г.) из Западной Европы [9].
С 30.07.2012 территория Украины официально неблагополучна по АЧС. За время эпизоотии (по данным Всемирной организации здравоохранения животных за 21.06.2024) зарегистрировано 620 случаев, из них 487 очагов среди домашних свиней и 133 – среди популяций дикого кабана (рис. 1)2. При этом не существует достоверной информации о циркуляции вируса АЧС на Украине и в регионах левобережья Днепра (в Донецкой, Луганской и Запорожской областях). Единственный штамм с установленной нуклеотидной последовательностью генома, выделенный 11.04.2016 (Kyiv/131 2016) в Киевской области Украины, описан Г. Коваленко и соавт. (2019 г.) и причислен к кластеру, характерному для образцов, выявленных на территории Польши [10].

Рис. 1. Распространение АЧС на территории Украины (2012–2024 гг.).
Fig. 1. Spread of ASF in Ukraine (2012–2024).
Трудности в понимании молекулярной эволюции вируса АЧС генотипа II в Евразии (2007 г. – наст. время), прежде всего, связаны с недостатком данных секвенирования, что также лимитирует прикладное значение филогенетического и пространственно-временного анализов в ветеринарии. В связи с этим молекулярно-генетические исследования, посвященные поиску новых и подтверждению циркуляции уже охарактеризованных геновариантов вируса во всех административных единицах евроазиатских стран, являются актуальным направлением научных исследований.
Цели исследования – осуществить полногеномное секвенирование и анализ олиго- и однонуклеотидного полиморфизма (ОНП) вируса АЧС, выделенного из биологического материала от домашних свиней, содержавшихся в Донецком, Луганском и Запорожском регионах в 2023 г.; идентифицировать уникальные точечные мутации, позволяющие дифференцировать генетические варианты; установить филогенетическое родство исследуемых изолятов и их принадлежность к субгенотипам.
Материалы и методы
Изоляты вируса АЧС. Образцы патологического материала от павших домашних свиней (мышечная ткань, селезенка, костный мозг) на территории Донецкого, Луганского и Запорожского субъектов направлялись в референтную лабораторию по АЧС (ФГБУ «ВНИИЗЖ», Владимир, Россия) с целью подтверждения лабораторного диагноза на АЧС и проведения научно-исследовательской работы (табл. 1). После положительного результата полимеразной цепной реакции в реальном времени из образцов готовили 10% гомогенизированную суспензию, которую использовали для выделения вируса. Идентификацию и накопление изолятов проводили в первичной культуре клеток костного мозга свиньи (КМС) по ранее опубликованному протоколу в титре не менее 6,0 lg ГАдЕ50/см3 [11].
Таблица 1. Краткие характеристики образцов, использованных в исследовании
Таble 1. Brief characteristics of samples used in the study
Наименование изолята Isolate name | Дата регистрации вспышки Date of outbreak registration | Место отбора образцов Place of sample collection | Титр вируса в КМС к 3-му пассажу, (lg ГАдЕ50/см3 ± SD) Viral load in PBM cells by 3rd passage (log10 HAD50/ml ± SD) |
ASFV/DNR/DP2023/2466-1 | 22.09.2023 | Донецкий регион, г. Волноваха Donetsk region, Volnovakha | 8,62 ± 0,21 |
ASFV/DNR/DP2023/2466-3 | 21.09.2023 | Донецкий регион, Волновахский район, с. Ивановка Donetsk region, Volnovakha district, Ivanovka village | 7,95 ± 0,14 |
ASFV/DNR/DP2023/3343-2 | 16.10.2023 | Донецкий регион, Тельмановский район, п. Андреевка Donetsk region, Telmanovsky district, Andreevka settlement | 7,58 ± 0,14 |
ASFV/Zaporozskaya/DP2023/ 2896-5 | 01.11.2023 | Запорожская область, Бердянский район, с. Чернигово-Токмачанск Zaporozhye region, Berdyansk district, Chernigovo-Tokmachansk settlement | 8,81 ± 0,21 |
ASFV/LNR/DP2023/42-1 | 26.12.2023 | Луганский регион, Старобельский район, с. Подгоровка Luhansk region, Starobelsky district, Podgorovka village | 7,0 ± 0,14 |
Полногеномное ресеквенирование. Пробоподготовку образцов геномной ДНК вируса АЧС (гДНК) и оценку ее качества осуществляли в соответствии с методическими рекомендациями по очистке, концентрированию и выделению генома вируса АЧС и каприпоксвирусов для полногеномного секвенирования3.
Очистку и концентрирование выполняли способом № 1 (среднескоростным центрифугированием при 4 °С и 7000 об/мин в течение 16 ч).
Приготовление библиотеки проводили с использованием набора MGIEasy Universal DNA Library Prep Set (MGI Tech, Китай). Высокопроизводительное секвенирование (HTS) выполняли на платформе DNBSEQ-G400 (MGI Tech) [12].
Сборка и анализ последовательностей. Сборку консенсусной последовательности выполняли методом картирования прочтений на референс-геном штамма Georgia 2007/1 (NC_044959.2) с определением контигов в программе Geneious Prime (2024.0.5). Для прогнозирования открытых рамок считывания (ОРС) и аннотации генома использовали утилиту Genome Annotation Transfer Utility (GATU) на платформе Java v. 8 [13]. Множественное выравнивание полученных последовательностей с импортированными из базы данных GenBank (приложение) и анализ нуклеотидных изменений (ОНП) проводили с применением алгоритма CLUSTAL W в программе Geneious Prime; трансляцию и выявление аминокислотных изменений – в программе SnapGene v. 5.2.1. Уровень гомологии оценивали на онлайн-платформе NCBI: Nucleotide BLAST. Построение дендрограммы осуществляли в программе Mega X по рекомендованной модели T92 + G + I (BIC = 537644,659; AICc = 535583,3783) методом максимального правдоподобия (Maximum Likelihood) с Bootstrap 100 итерациями начальной загрузки [14].
Субгенотипирование изолятов проводили согласно методическим рекомендациям по молекулярно-эпизоотологической кластеризации вируса АЧС методом субгенотипирования (модификация классификации субгенотипов C. Gallardo и соавт., 2023)4 [15].
Этическое утверждение. Авторы подтверждают соблюдение институциональных и национальных стандартов по использованию лабораторных животных в соответствии с Consensus author guidelines for animal use (IAVES 23 July 2010). Протокол исследования одобрен Комиссией по биоэтике ФГБУ «ВНИИЗЖ» (протокол от 15.05.2024 № Dnieper/2024).
Результаты
Оценка качества гДНК и ресеквенирования. Определены спектрофотометрические показатели (отношение поглощений на длинах волн А260/А230 и А260/А280), а также биоинформатические метаданные, представленные на графиках (рис. 2).

Рис. 2. Спектрофотометрические показатели (а) образцов гДНК и биоинформатические метаданные: число прочтений (б), специфичность прочтений (в), покрытие (г), полученные после ресеквенирования.
Примечание: области с низким покрытием обозначены . – ASFV/DNR/DP2023/2466-1; 2 – ASFV/DNR/DP2023/2466-3; 3 – ASFV/DNR/DP2023/3343- 2; 4 – ASFV/Zaporozskaya/DP2023/2896-5; 5 – ASFV/LNR/DP2023/42-1.
Fig. 2. Spectrophotometric indices (a) of gDNA samples and bioinformatic metadata: number of reads (b), specificity of reads (c), coverage (d) obtained after resequencing.
Note: fragments with l ow coverage are indicated . 1 – ASFV/DNR/DP2023/2466-1; 2 – ASFV/DNR/DP2023/2466-3; 3 – ASFV/DNR/DP2023/3343-2; 4 – ASFV/Zaporozskaya/DP2023/2896-5; 5 – ASFV/LNR/DP2023/42-1.
Как показано на рис. 2, все образцы имели удовлетворительные показатели очистки гДНК от низкомолекулярных соединений, так как А230/А260 ≥ 2,0. Однако показатель А260/А280 составлял 1,77–1,80, что меньше рекомендуемого значения (≥ 1,8) и указывает на недостаточную чистоту от белковых примесей. В свою очередь, доля специфичных прочтений находилась в диапазоне от 0,69 до 1,14%, а средняя глубина покрытия – 407–1664 прочтений/нуклеотид, превышая рекомендуемое значение (> 30). Область с низким покрытием у 3 изолятов из Донецкого региона находилась в позициях 187,750 до 189,000 т.п.н. В то время как у ASFV/Zaporozskaya/DP2023/2896-5 – в положении 16,640–18,430 т.п.н. При картировании прочтений на референс-геном штамма Georgia 2007/1 (190,584 т.п.н.) сформировался один длинный контиг в отсутствии скаффолдов для каждого изолята, впоследствии составивший консенсусные последовательности длиной от 190,573 до 190,596 т.п.н.
Аналогично штамму Georgia 2007/1 у всех исследуемых геномов идентифицировано 195 ОРС.
По результатам сборки и аннотации генома последовательности депонированы в базу данных VGARus под идентификационными номерами niiz000001-5.
Анализ ОНП. Установлена принадлежность всех изученных изолятов к генотипу II на основании С-терминального фрагмента гена B646L.
Множественное полногеномное выравнивание показало наличие одно- и олигонуклеотидного полиморфизма. Отмечены уникальные (вновь выявленные) и охарактеризованные синонимичные и несинонимичные замены, инделы, представленные в табл. 2.
Таблица 2. Сравнительный анализ точечных мутаций и аминокислотных изменений
Table 2. Comparative analysis of point mutations and amino acid changes
Нуклеотидная позиция Nucleotide position (Georgia 2007/1) | Характер ОНП Type of SNP | Аминокислотная изменчивость Variability of amino acid | Ген/Межгенная область Gene/Intergenic region | Встречаемость Occurrence |
1572 | T → C | – | Межгенная область Intergenic region | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
1587 | T → C | – | Межгенная область Intergenic region | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
1611 | A → Т | – | Межгенная область Intergenic region | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 |
2817 | A → Т | L → stop | MGF 360-1Lb | ASFV/Zaporozskaya/DP2023/2896-5 |
2929 | Делеция / Deletion A | – | Межгенная область / Intergenic region 1L/2L | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
7059 | C → T | W → stop | MGF 110-1L | Все исследуемые изоляты, за исключением All of the isolates tested except Ulyanovsk/19/WB/5699 Kabardino-Balkaria/19/WB/ 964 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 |
8280 | G → А | H → Y | MGF 110-3L | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 |
8329 | T → C | G = G | MGF 110-3L | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 |
9253 | C → А | V → L | MGF 110-4L | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
16283 | A → G | – | PolyC-region | ASFV/LNR/DP2023/42-1 |
16683 | C → T | A = A | MGF 360-4L | ASFV/Zaporozskaya/DP2023/2896-5 |
16692 | A → G | V → I | ||
16694 | C → T | I → V | ||
16703 | T → C | I → V | ||
16718 | T → C | V → I | ||
16719 | G → А | A = A | ||
16727 | G → T | Q → K | ||
16738 | A → Т | L → Y | ||
16739 | G → А | L → Y | ||
16745 | A → G | L = L | ||
16746 | A → G | H = H | ||
17509 | A → Т | – | Межгенная область Intergenic region | ASFV/LNR/DP2023/42-1 |
18576 | A → G | A = A | MGF 360-6L | ASFV/Zaporozskaya/DP2023/2896-5 |
18582 | C → А | S → F | ||
18583 | G → А | S → F | ||
18592 | T → A | D → V | ||
18594 | A → G | N = N | ||
18598 | G → А | P → L | ||
18600 | A → C | D → G | ||
18601 | T → C | D → G | ||
24690 | G → А | T → M | MGF 360-8L | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
24967 | A → G | S → P | MGF 360-8L | ASFV/Zaporozskaya/DP2023/2896-5 |
30904 | C → T | L = L | MGF 360-12L | ASFV/Zaporozskaya/DP2023/2896-5 |
44576 | A → G | K → E | MGF 505-9R | Все исследуемые изоляты, за исключением All of the isolates tested except Ulyanovsk/19/WB/5699 Kabardino-Balkaria/19/WB/ 964 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 |
46135 | C → А | F → L | MGF 505-10R | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
46557 | G -> А | R → Q | MGF 505-10R | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
48231 | Инсерция Insertion CTAGCTATAG | – | Межгенная область Intergenic region | ASFV/Zaporozskaya/DP2023/2896-5 |
49085 | T → A | N → Y | A240L | ASFV/LNR/DP2023/42-1 |
50655 | G → А | A → T | MGF 360-15R | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
50667 | G → А | E → K | MGF 360-15R | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
54369 | C→ T | S → N | A859L | ASFV/LNR/DP2023/42-1 |
54758 | G → А | – | Межгенная область Intergenic region | ASFV/Zaporozskaya/DP2023/2896-5 |
57427 | G → А | H = H | F334L | ASFV/Zaporozskaya/DP2023/2896-5 |
74708 | A → G | T → A | EP402R | ASFV/DNR/DP2023/3343-2 |
86659 | G → А | A = A | C257L | ASFV/Zaporozskaya/DP2023/2896-5 |
106942 | C → T | V → I | B117L | ASFV/LNR/DP2023/42-1 |
121744 | G → А | I = I | CP2475L | ASFV/LNR/DP2023/42-1 |
131463 | G → C | Q → E | NP1450L | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
134514 | T → C | R → S | NP419L | Все исследуемые изоляты, за исключением All of the isolates tested except Ulyanovsk/19/WB/5699 Kabardino-Balkaria/19/WB/ 964 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 |
157272 | G → А | A → V | R298L | ASFV/Zaporozskaya/DP2023/2896-5 |
157297 | G → А | A → V | R298L | ASFV/Zaporozskaya/DP2023/2896-5 |
158805 | C → G | E → Q | Q706L | ASFV/LNR/DP2023/42-1 ASFV/Bryanskaya 2021/DP-18 ASFV/Bryanskaya 2021/DP-8823 |
167062 | C → T | G → R | I199L | ASFV/Zaporozskaya/DP2023/2896-5 ASFV/Kaliningrad 18/WB-9767 ASFV/Kaliningrad 18/WB-12524 ASFV/Kaliningrad 18/WB-9766 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 |
167188 | C → G | A → P | ASFV/DNR/DP2023/2466-1, ASFV/Zabaykali 2020/WB-5314 ASFV/Zabaykaly_2020/DP-4905 | |
167196 | G → А | T → I | ASFV/LNR/DP2023/42-1 | |
168627 | T → A | F → I | E248R | ASFV/LNR/DP2023/42-1 |
170862 | T → A | I → F | I267L | Все исследуемые изоляты, за исключением All of the isolates tested except Ulyanovsk/19/WB/5699 Kabardino-Balkaria/19/WB/ 964 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 Odintsovo/WB/Russia/2014 |
173273 | C → T | T = T | I73R | ASFV/LNR/DP2023/42-1 |
173408 | Инсерция Insertion GGAATATATA | – | Межгенная область Intergenic region I73R/I329L | 27 из 45 исследуемых изолятов, включая 5 описанных в настоящем исследовании 27 of the 45 isolates investigated, including the 5 described in this study |
184404 | G → А | A → T | MGF 360-18R | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
187684 | A → C | – | Межгенная область Intergenic region | ASFV/DNR/DP2023/2466-1 ASFV/DNR/DP2023/2466-3 ASFV/DNR/DP2023/3343-2 |
187700 | A → Т | – | ||
187701 | G → А | – | ||
187702 | G → T | – | ||
187703 | G → А | – | ||
187718 | T → C | – | ||
190116 | Инсерция Insertion А | I → N | DP60R | Все исследуемые изоляты, за исключением All of the isolates tested except Ulyanovsk/19/WB/5699 Kabardino-Balkaria/19/WB/ 964 Pol15/Podlaskie/Poland//2015 LT14/1490/Lithuania/2014 Odintsovo/WB/Russia/2014 ASFV CzechRepublic 2017/1 ASFV Germany 2020/1 |
Таким образом, у 5 изолятов вируса АЧС выявлено 63 замены (44 транзиции и 19 трансверсий), 14 из которых оказались синонимичными и 38 – несинонимичными, приводящими к изменению аминокислотного состава кодируемого белка; 3 инсерции: 1 – в ОРС, 2 – в межгенных областях; 1 однонуклеотидная делеция в межгенной области.
Десять нуклеотидных замен оказались уникальны для изолятов из Донецкой области, 6 из них содержатся в локусах мультигенных семейств MGF 110, 360, 505. Несинонимичная транзиция A → G в гене EP402R, кодирующем ответственный за сероиммунотипирование гемадсорбирующий гликопротеин CD2v вируса АЧС, зарегистрирована исключительно у ASFV/DNR/DP2023/3343-2.
У вируса АЧС из Запорожской области отмечались многочисленные ранее неохарактеризованные замены в мультигенном семействе MGF 360 (6 трансверсий и 16 транзиций), которые значительно изменили аминокислотную последовательность одноименных белков. Две одинаковые несинонимичные транзиции G → А, повторяемые в гене R298L, привели к замене аланина (А) на валин (V).
Вирус АЧС, изолированный на территории Луганского региона (ASFV/LNR/DP2023/42-1), имел 9 уникальных замен и 6 ранее характерных только для двух образцов, выделенных от домашних свиней на крупных предприятиях Брянской области в 2021 г. (ASFV/Bryanskaya 2021/DP-18; ASFV/Bryanskaya 2021/DP-8823).
Все 5 исследуемых образцов принадлежали генетическим вариантам (II) с единичными заменами по маркерам I267L, NP419L, MGF 505-9R и MGF 110-1L. Во всех 4 генах на выравнивании наблюдались сочетанные мутации, за исключением штамма Odintsovo/WB/Russia/2014 (рис. 3).

Рис. 3. Выравнивание последовательностей генов MGF 110-1L (а), MGF 505-9R (б), NP419L (в) и I267L (г) с указанием несинонимичных замен.
Fig. 3. Multiple alignment of MGF 110-1L (a), MGF 505-9R (b), NP419L (c), and I267L (d) genes showing non-synonymous substitutions.
Анализ ОНП генов Е199L и DP60R не показал корреляции молекулярной эволюции вируса АЧС и пространственно-временного кластерного распространения в связи с регистрацией отличных от штамма Georgia 2007/1 генетических вариантов в отдаленных друг от друга географических областях. Так, замена C → T в позиции 167062 локуса Е199L и инсерция А в положении 190116 гена DP60R встречалась у ряда изолятов из Польши, Литвы, Китая, Германии, Чехии, а также из Калининградской области, Дальневосточного федерального округа и центральных регионов России.
Межгенная область I73R/I329L содержала три 10-нуклеотидных инсерций тандемных повторов (TRS) и относилась к IGR-II, превалирующему на территории Евразии.
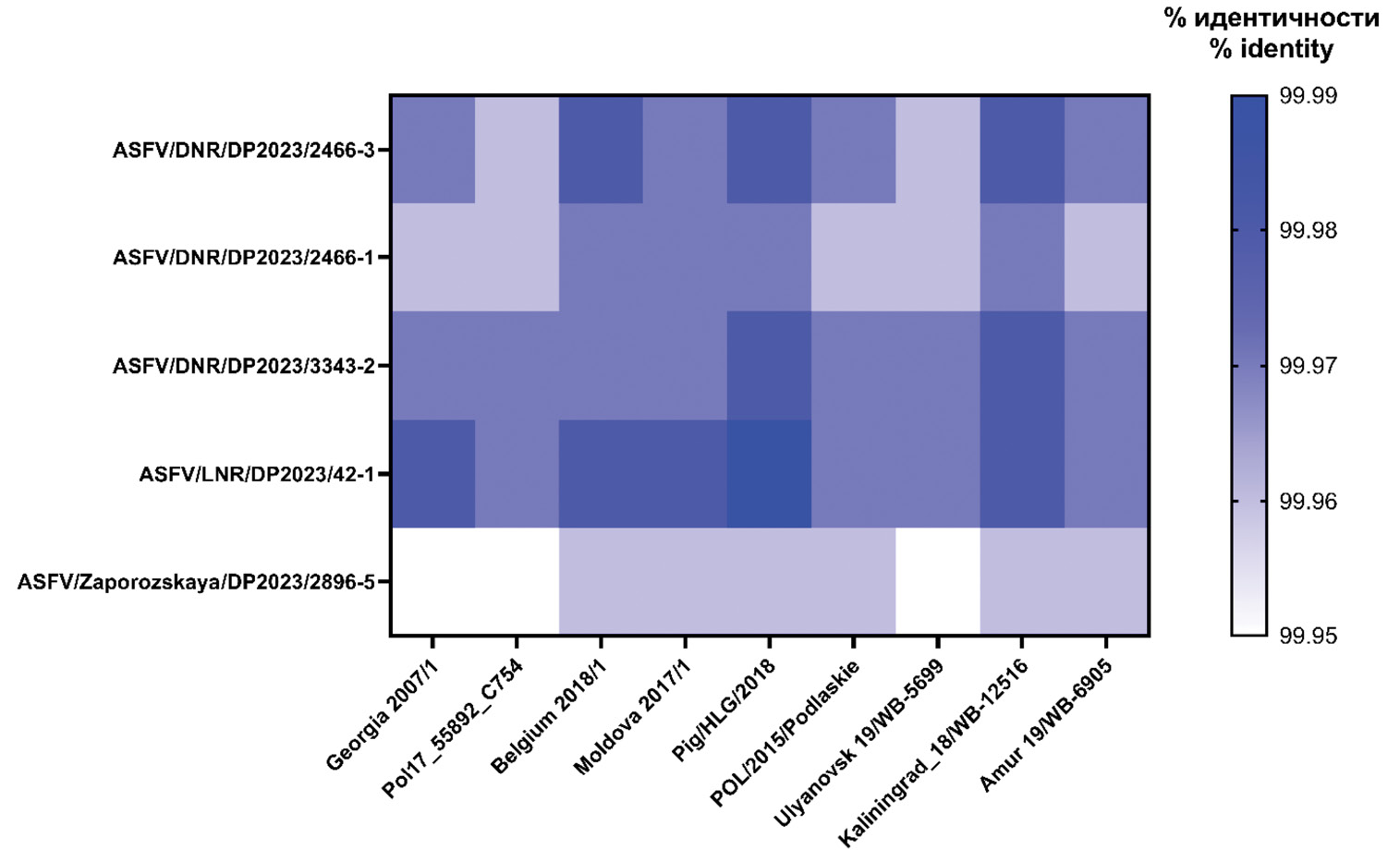
Уровень гомологии. Установлена гомологичность исследуемых полногеномных последовательностей с наиболее изученными (референсными) штаммами вируса АЧС, принадлежащими генотипу II и выделенными в разных энзоотичных странах Евразии (Россия: Калининградская («Запад»), Ульяновская («Центр») и Амурская («Восток») области; Молдова, Бельгия, Польша: 2 гетерогенных генетических варианта (до 2016 г. и после), Китай), при вычислении процента идентичности (рис. 4).

Рис. 4. Гомология изолятов, выделенных на территории левобережья Днепра, со штаммами, охарактеризованными в географически отдаленных друг от друга областях Евразии.
Fig. 4. Homology of isolates collected on the territory of the Dnieper left bank with strains characterized in geographically distant areas of Eurasia.
По данным рис. 4, циркулирующий вирус АЧС в Донецкой, Луганской и Запорожской областях обладает высоким генетическим сродством (99,95–99,99% идентичности) со штаммами II генотипа, выделенными на евроазиатском континенте в течении современной эпизоотии АЧС.
Филогенетический анализ. Определено филогенетическое родство 45 штаммов и изолятов вируса АЧС, импортированных из реестра GenBank, с исследуемыми последовательностями. Результаты анализа представлены на рис. 5.

Рис. 5. Филограмма изолятов вируса АЧС, выделенных в Евразии с 2007 по 2023 г.
Примечание: изученные в настоящем исследовании изоляты обозначены ●.
Fig. 5. Phylogenetic tree of ASFV isolates collected in Eurasia from 2007 to 2023.
Note: isolates obtained in this study are labeled ●.
На укорененной дендрограмме (рис. 5) идентифицируются три крупные группы изолятов, ветви которых имеют один общий внутренний узел. Изученные геноварианты можно разделить на 6 клад (кластеров) ввиду значительной как филогенетической, так и географической обособленности. Так, изоляты исходной (корневой) клады обозначены синим цветом и наименованы как «Грузия 2007», поскольку характеризуются более ранним происхождением и высоким родством с материнским штаммом Georgia 2007/1. Бинарный узел на дереве разделяет вирус АЧС, циркулирующий в европейских странах, на две клады, получивших названия «Восточная Европа» (Калининградская область, Польша, Германия, выделена красным цветом) и «Европа» (Молдова, Чехия, Бельгия, отмечена оранжевым цветом). От второго крупного узла отходят три сестринские клады изолятов: «Азия» (Китай и Дальний Восток России показана желтым цветом), «Центр России 2021» (Свердловская и Белгородская области, Пермский край – обозначена темно-зеленым оттенком) и «Брянск 2021» (Брянская область – имеет светло-зеленый оттенок).
ASFV/LNR/DP2023/42-1 проявляет филогенетическое родство и относится к одной группе с вирусом АЧС из Брянской области (20% ‒ 1/5), вызывавшим вспышки АЧС на свиноводческих комплексах в 2021 г.
Последовательности вируса АЧС, изолированного в Донецком и Запорожском регионах (80% ‒ 4/5), группируются в кладу «Европа». При этом все три изолята из Донецкой области формируют собственный внутренний субкластер. Длина ветви ASFV/Zaporozskaya/DP2023/2896-5 гораздо больше остальных последовательностей, что указывает на высокое число добавочных замен.
Все изоляты II генотипа, выделенные в странах Евразии, принадлежат монофилетической группе, т.е. имеют общего предка (вероятно, штамм Georgia 2007/1).
Субгенотипирование. На основании распределения генетических вариантов по 9 маркерным фрагментам генома вируса АЧС идентифицирован субгенотип (геногруппа; табл. 3).
Таблица 3. Данные субгенотипирования изолятов вируса АЧС, исследованных в работе
Table 3. Subgenotyping data of ASFV isolates studied in this research
Характеристики Features | Изолят Isolate | ||||
ASFV/DNR/ DP2023/2466-1 | ASFV/DNR/ DP2023/2466-3 | ASFV/DNR/ DP2023/3343-2 | ASFV/Zaporozskaya/ DP2023/2896-5 | ASFV/LNR/ DP2023/42-1 | |
CVR | I | I | I | I | I |
IGR I73R/I329L | II | II | II | II | II |
MGF 505-9R/10R | I | I | I | I | I |
K145R | I | I | I | I | I |
O174L | I | I | I | I | I |
MGF 505-5R | I | I | I | I | I |
MGF 360-10L | I | I | I | I | I |
I267L | II | II | II | II | II |
I215L | I | I | I | I | I |
Генотип Genotype | II | II | II | II | II |
Cубгенотип Subgenotype | 3b | 3b | 3b | 3b | 3b |
Все 5 изученных изолятов относятся к субгенотипу 3b, получившему повсеместное распространение на территории всех регионов страны, за исключением Калининградской области и Дальневосточного федерального округа. В сравнительном анализе 45 изолятов к геногруппе 3b относились 13 (28,9%: исследуемые, Belgorodskaya 2021/DP-11838, Bryanskaya 2021/DP-8823, Bryanskaya 2021/DP-18, Permskyi 2021/DP-9916, Sverdlovskaya 2021/DP-9914, Belgium 2018/1, Moldova 2017/1, CzechRepublic 2017/1), характерных для Европы и России в период с 2017 по 2023 г.
Обсуждение
До настоящего момента данные полногеномного анализа вируса АЧС, изолированного на территории левобережья Днепра, в открытой печати отсутствовали. В данном исследовании впервые приведены результаты выявления точечных мутаций и филогении у 5 изолятов, выделенных в Донецкой, Луганской и Запорожской областях в 2023 г.
На примере последовательностей, импортированных из GenBank, показано формирование клад «Грузия 2007» (референс, характерный для начала эпизоотии АЧС в период с 2007 по 2015 г. и спорадически регистрируемый до 2019 г.), «Центр России 2021», «Брянск 2021», «Азия», «Европа» и «Восточная Европа» [5–9]. Установлена принадлежность 4 изолятов из Донецкого и Запорожского регионов к кластеру «Европа», что предполагает независимую и непрерывную молекулярную эволюцию этого геноварианта вируса АЧС II генотипа на территории как западно- (Бельгия), так и восточноевропейских (Чехия, Молдова, Украина) стран как минимум с 2017 по 2023 г. (рис. 5).
Подтверждением втохтонности также служит распределение аллелей по маркерным нуклеотидным участкам в субгенотип 3b, наиболее распространенный в странах Европы и Центральных субъектах России с 2016 г. Стоит обратить внимание, что изоляты кластеров «Европа», «Центр России 2021» и «Брянск 2021» относятся к одному субгенотипу (3b), а клады «Азия» – к другим (3d и 3i), несмотря на результаты полногеномного анализа, показывающие близкое родство «центральных» и «азиатских» вариантов [15]. Слабую корреляцию методов можно обосновать различными приоритетами исследования, т.к. филогения полных последовательностей генома определяет главным образом показатели происхождения и степени дивергенции вируса, тогда как фрагментарный («локальный») анализ – циркуляцию возбудителя в определенной физико-географической/административной зоне.
Штамм Kyiv/131 2016 при этом не был включен в сравнительный анализ из-за обнаруженного большого числа ошибок секвенирования/сборки генома, способствующих формированию недостоверных выводов [10]. Однако геноварианты маркерных областей генома Kyiv/131 2016 идентичны изолятам, относящимся к кластеру «Восточная Европа» [16].
Таким образом, вспышки АЧС у домашних свиней, содержащихся на правобережной и левобережной территории Днепра, были вызваны вирусом АЧС, принадлежащим разным субгенотипам (7 и 3b соответственно). В свою очередь, пространственно-временной анализ значительно лимитирован по причине недостаточных молекулярно-эпизоотологических данных о распространении АЧС на Украине в период с мая 2016 по август 2023 г.
Высокий уровень гомологии (99,95–99,99%) охарактеризованных изолятов с другими штаммами II генотипа, выделенными в странах Евразии (Грузии, Китае, Польше, Молдовы, Бельгии и России), в очередной раз подтверждают генетическую консервативность вируса АЧС, инфицирующего домашних свиней и диких кабанов (один вид – Sus scrofa) и дивергентного от родительского варианта «Грузия 2007» [6]. Следует отметить, что такие заключения применимы исключительно к вирусу АЧС II генотипа и не коррелируют с сообщениями о распространении возбудителя I генотипа или рекомбинантного варианта в Китайской Народной Республике [17, 18].
Большинство замен (6/10) у изолятов, выделенных в Донецком регионе, расположены в мультигенных семействах MGF 110, 360, 505, которые могут кодировать факторы вирулентности вируса АЧС [19]. ОНП в локусе EP402R у изолята ASFV/DNR/DP2023/3343-2 может приводить к изменению сероиммуноспецифичности, что необходимо подтверждать в иммунопробе на животных и в реакции задержки гемадсорбции (РЗГАд) с использованием референс-сывороток [20].
Изолят ASFV/Zaporozskaya/DP2023/2896-5 обладает выраженным полиморфизмом генов MGF 360, проявляющимся в наличии 22 замен, 15 из которых несинонимичные и 7 – синонимичные. Выявленный ОНП служит основанием для проведения экспериментальных работ по оценке иммунобиологических свойств на естественно-восприимчивых животных, т.к. мутагенез в генах MGF может приводить к снижению вирулентных свойств вируса [21].
Филогенетический анализ полногеномных последовательностей показал родство изолятов, выделенных в Луганском и Брянском субъекте (2021 г., рис. 5). Так, 6/9 замен, выявленных у ASFV/LNR/DP2023/42-1, идентичны ранее уникальным для вируса АЧС из Брянской области, однако 3/9 замен обнаружены впервые, что может указывать на происхождение генетического варианта от исходного, близкого кладе «Брянск 2021».
Следует отметить, что все 5 изученных изолятов на филогенетической дендрограмме имели длину ветви, значительно превосходящую этот параметр у исходных кладоформирующих штаммов, что указывает на большее число дополнительных ОНП (мутационный темп в заменах/сайт/год) и гипотетически на более позднее происхождение геновариантов при условии корректного секвенирования (рис. 5). В этом отношении результаты филогении подтверждаются известными эпизоотологическими данными о вспышках АЧС, представленными в работе (табл. 1).
Исследование полиморфизма генов E199L и DP60R у 45 последовательностей генотипа II, включая представленные впервые, выявило отсутствие подтверждающих перспектив использования этих локусов в качестве маркерных фрагментов генома для отслеживания распространения АЧС в Евразии, в отличие от предыдущих данных [7]. Тождественный ОНП у изолятов из географически отдаленных регионов может быть связан с наличием у вируса АЧС точечной репарации генома, обеспечивающегося за счет функционирования Pol X ДНК-полимеразы, поскольку E199L кодирует важнейший белок, связанный с активацией аутофагии зараженных клеток [22, 23]. В любом случае, данные анализа генов E199L и DP60R, опровергающие энзоотичность АЧС, противоречат результатам исследования прочих фрагментов, включая маркерных, подтверждающих территориально-ограниченное развитие эпизоотического процесса.
Кроме того, не обнаружено ни одной точечной мутации, которая была бы исключительна для всех изолятов, выделенных на территории левобережья Днепра в 2023 г., что указывает на независимые пути генетической изменчивости вируса АЧС, инфицирующего восприимчивых животных в этих субъектах. Однако выявлен ряд уникальных ОНП для каждого охарактеризованного изолята, позволяющий в дальнейшем идентифицировать родственные геноварианты с описанными в настоящем исследовании. Разработка праймеров, фланкирующих фрагменты с выявленными специфичными мутациями, поможет ускорить процесс дифференциации в дальнейших работах.
Заключение
Впервые проведен полногеномный анализ вируса АЧС, циркулирующего на территории левобережья Днепра, на примере 5 изолятов, выделенных в Донецкой, Луганской и Запорожской областях от домашних свиней в 2023 г. Все они отнесены к генотипу II, имеют монофилетическое происхождение, филогенетически близки кладам «Европа» (4/5) и «Брянск 2021» (1/5).
В целом на территории данных субъектов не отмечается эмерджентность АЧС, а характерно внутрикластерное распространение. Изученные изоляты относятся к субгенотипу 3b и имеют высокий уровень гомологии (99,95–99,99%) с большинством референтных штаммов, принадлежащих генотипу II и зарегистрированных в Евразии. Однако выявлены ранее не описанные точечные мутации, уникальные для каждого изолята, которые позволят идентифицировать родственные варианты.
Кроме того, обнаружены многочисленные замены в локусах мультигенного семейства MGF 110, 505 и 360, кодирующих факторы вирулентности, у 4 изолятов из Донецкой и Запорожской областей, что может также влиять на изменение иммунобиологических свойств возбудителя АЧС.
На примере изучения филогении показаны достаточные для дифференциации показатели вариабельности и гетерогенности вируса АЧС генотипа II, происходящего от референс-штамма Georgia 2007/1. Представленные данные обладают теоретической и практической значимостью при усовершенствовании и разработке мер профилактики, а также могут значительно расширить возможности внутреннего и международного надзора за АЧС.
1 Россельхознадзор. Эпизоотическая ситуация по АЧС на территории Российской Федерации, в странах Европы, Азии и Америки. Данные ВОЗЖ с 2007 по 2023 г.; 2023. Available at: https://fsvps.gov.ru/wp-content/uploads/2023/06/05_АЧС_2007_2023_мир.png
2 Россельхознадзор. Эпизоотическая ситуация по АЧС на территории Украины; 2024. Available at: https://fsvps.gov.ru/wp-content/uploads/2023/06/АЧС-в-Украине-17.pdf
3 Мазлум А., Чернышев Р.С., Кротова А.О. и др. Методические рекомендации по очистке, концентрированию и выделению генома вируса африканской чумы свиней и каприпоксвирусов для полногеномного секвенирования. Владимир; 2024.
4 Чернышев Р.С., Мазлум А., Зиняков Н.Г. и др. Методические рекомендации по молекулярно-эпизоотологической кластеризации изолятов вируса африканской чумы свиней методом субгенотипирования. Владимир; 2024.
Об авторах
Роман Сергеевич Чернышев
ФГБУ «Федеральный центр охраны здоровья животных»
Автор, ответственный за переписку.
Email: chernishev_rs@arriah.ru
ORCID iD: 0000-0003-3604-7161
аспирант, ветеринарный врач референтной лаборатории по африканской чуме свиней
Россия, 600901, г. ВладимирАлексей Сергеевич Иголкин
ФГБУ «Федеральный центр охраны здоровья животных»
Email: igolkin_as@arriah.ru
ORCID iD: 0000-0002-5438-8026
канд. вет. наук, заместитель руководителя центра – заведующий референтной лабораторией по африканской чуме свиней лабораторно-диагностического центра
Россия, 600901, г. ВладимирНиколай Геннадьевич Зиняков
ФГБУ «Федеральный центр охраны здоровья животных»
Email: zinyakov@arriah.ru
ORCID iD: 0000-0002-3015-5594
канд. биол. наук, ведущий научный сотрудник референтной лаборатории вирусных болезней птиц
Россия, 600901, г. ВладимирИлья Александрович Чвала
ФГБУ «Федеральный центр охраны здоровья животных»
Email: chvala@arriah.ru
ORCID iD: 0000-0002-1659-3256
канд. вет. наук, заместитель директора
Россия, 600901, г. ВладимирСписок литературы
- Beltrán-Alcrudo D., Lubroth J., Depner K., Rocque S. African swine fever in the Caucasus. EMPRES Watch. 2008; 1(8): 1–8. https://doi.org/10.13140/RG.2.1.3579.1200
- Nix R.J., Gallardo C., Hutchings G., Blanco E., Dixon L.K. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch. Virol. 2006; 151(12): 2475–94. https://doi.org/10.1007/s00705-006-0794-z
- Shen Z.J., Jia H., Xie C.D., Shagainar J., Feng Z., Zhang X., et al. Bayesian phylodynamic analysis reveals the dispersal patterns of African swine fever virus. Viruses. 2022; 14(5): 889. https://doi.org/10.3390/v14050889
- Malogolovkin A., Yelsukova A., Gallardo C., Tsybanov S., Kolbasov D. Molecular characterization of African swine fever virus isolates originating from outbreaks in the Russian Federation between 2007 and 2011. Vet. Microbiol. 2012; 158(3-4): 415–9. https://doi.org/10.1016/j.vetmic.2012.03.002
- Chapman D.A., Darby A.C., Da Silva M., Upton C., Radford A.D., Dixon L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011; 17(4): 599–605. https://doi.org/10.3201/eid1704.101283
- Mazloum A., van Schalkwyk A., Shotin A., Igolkin A., Shevchenko I., Gruzdev K.N., et al. Comparative analysis of full genome sequences of African swine fever virus isolates taken from wild boars in Russia in 2019. Pathogens. 2021; 10(5): 521. https://doi.org/10.3390/pathogens10050521
- Чернышев Р.С., Спрыгин А.В., Шотин А.Р., Иголкин А.С., Мазлум А. Сравнительный анализ полногеномных последовательностей двух изолятов вируса африканской чумы свиней, выделенных в 2020 году от домашних и диких свиней в Забайкальском крае Российской Федерации. Ветеринария, зоотехния и биотехнология. 2022; (10): 84–97. https://doi.org/10.36871/vet.zoo.bio.202210010 https://elibrary.ru/qcgsux
- Zhang Y., Wang Q., Zhu Z., Wang S., Tu S., Zhang Y., et al. Tracing the origin of genotype II African swine fever virus in China by genomic epidemiology analysis. Transbound. Emerg. Dis. 2023; (1): 4820809. https://doi.org/10.1155/2023/4820809
- Xin G., Kuang Q., Le S., Wu W., Gao Q., Gao H., et al. Origin, genomic diversity and evolution of African swine fever virus in East Asia. Virus Evol. 2023; 9(2): vead060. https://doi.org/10.1093/ve/vead060
- Kovalenko G., Ducluzeau A.L., Ishchenko L., Sushko M., Sapachova M., Rudova N., et al. Complete genome sequence of a virulent African swine fever virus from a domestic pig in Ukraine. Microbiol. Resour. Announc. 2019; 8(42): e00883–19. https://doi.org/10.1128/MRA.00883-19
- Puzankova O., Gavrilova V., Chernyshev R., Kolbin I., Igolkin A., Sprygin A., et al. Novel protocol for the preparation of porcine bone marrow primary cell culture for African swine fever virus isolation. Methods Protoc. 2023; 6(5): 73. https://doi.org/10.3390/mps6050073
- Sun X., Hu Y.H., Wang J., Fang C., Li J., Han M., et al. Efficient and stable metabarcoding sequencing data using a DNBSEQ-G400 sequencer validated by comprehensive community analyses. GigaByte. 2021; 2021: gigabyte16. https://doi.org/10.46471/gigabyte.16
- Tcherepanov V., Ehlers A., Upton C. Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Genomics. 2006; 7: 150. https://doi.org/10.1186/1471-2164-7-150.
- Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018; 35(6): 1547–9. https://doi.org/10.1093/molbev/msy096
- Gallardo C., Casado N., Soler A., Djadjovski I., Krivko L., Madueño E., et al. A multi gene-approach genotyping method identifies 24 genetic clusters within the genotype II-European African swine fever viruses circulating from 2007 to 2022. Front. Vet. Sci. 2023; (10): 1112850. https://doi.org/10.3389/fvets.2023.1112850
- Чернышев Р.С., Иголкин А.С., Шотин А.Р., Зиняков Н.Г., Колбин И.С., Садчикова А.С. и др. Пространственно-временной кластерный анализ циркуляции вируса африканской чумы свиней (Asfarviridae: Asfivirus) в Калининградской области на основе трех генетических маркеров. Вопросы вирусологии. 2024; 69(3): 241–54. https://doi.org/10.36233/0507-4088-231 https://elibrary.ru/lbevpz
- Sun E., Huang L., Zhang X., Zhang J., Shen D., Zhang Z., et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg. Microbes Infect. 2021; 10(1): 2183–93. https://doi.org/10.1080/22221751.2021.1999779
- Zhao D., Sun E., Huang L., Ding L., Zhu Y., Zhang J., et al. Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs. Nat. Commun. 2023; 14(1): 3096. https://doi.org/10.1038/s41467-023-38868-w
- Zhu Z., Chen H., Liu L., Cao Y., Jiang T., Zou Y., et al. Classification and characterization of multigene family proteins of African swine fever viruses. Brief. Bioinform. 2021; 22(4): bbaa380. https://doi.org/10.1093/bib/bbaa380
- Балышев В.М., Болгова М.В., Балышева В.И., Болгова М.В., Князева М.В., Живодеров С.П. Получение типовых задерживающих гемадсорбцию референс-сывороток к вирусу африканской чумы свиней. Вопросы нормативно-правового регулирования в ветеринарии. 2015; (2): 23–5. https://elibrary.ru/twnfvt
- Wu L., Yang B., Yuan X., Hong J., Peng M., Chen J.L., et al. Regulation and evasion of host immune response by African swine fever virus. Front. Microbiol. 2021; 12: 698001. https://doi.org/0.3389/fmicb.2021.698001
- Redrejo-Rodríguez M., Rodríguez J.M., Suárez C., Salas J., Salas M.L. Involvement of the reparative DNA polymerase Pol X of African swine fever virus in the maintenance of viral genome stability in vivo. J. Virol. 2013; 87(17): 9780–7. https://doi.org/10.1128/JVI.01173-13
- Chen S., Zhang X., Nie Y., Li H., Chen W., Lin W., et al. African swine fever virus protein E199L promotes cell autophagy through the interaction of PYCR2. Virol. Sin. 2021; 36(2): 196–206. https://doi.org/10.1007/s12250-021-00375-x
Дополнительные файлы

