



Клеточные белки – потенциальные мишени антиретровирусной терапии
- Авторы: Бобкова М.Р.1
-
Учреждения:
- ФГБНУ «Научно-исследовательский институт вакцин и сывороток им. И.И. Мечникова»
- Выпуск: Том 68, № 6 (2023)
- Страницы: 488-504
- Раздел: ОБЗОРЫ
- URL: https://virusjour.crie.ru/jour/article/view/16597
- DOI: https://doi.org/10.36233/0507-4088-207
- EDN: https://elibrary.ru/klgwak
- ID: 16597

Цитировать

Аннотация
Обзорная статья содержит анализ информации, полученной в результате поиска литературы по базам данных Scopus, Web of Science, MedLine. Тема поиска – идентификация и изучение механизмов действия факторов хозяйской клетки, участвующих в цикле репликации вируса иммунодефицита человека (ВИЧ, Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodeficiency virus-1). Приведены примеры двух основных групп белков – факторов зависимости ВИЧ (CypA, LEDGF, TSG101 и др.) и факторов рестрикции (SERINС5, TRIM5α, APOBEC3G и др.); описано современное состояние представлений о механизмах их функционирования. Дана оценка перспектив разработки лекарственных средств для лечения ВИЧ-инфекции, направленных на ингибирование либо стимуляцию активности хозяйских факторов.
Ключевые слова
Полный текст
Успехи антиретровирусной терапии (АРТ), основанной на применении препаратов, непосредственно воздействующих на ферменты вируса, радикально изменили лицо эпидемии ВИЧ-инфекции, превратив ее из неминуемо смертельной в контролируемую с восстановлением срока и качества жизни инфицированных людей. Несмотря на этот замечательный прогресс, все проблемы АРТ решить не удается до сих пор, спустя почти 30 лет с начала внедрения эффективных схем тритерапии препаратами прямого действия. Главные из этих проблем: 1) невозможность полного излечения ВИЧ-инфекции вследствие феномена латентности, лежащего в основе патогенеза этого заболевания; 2) как следствие, необходимость пожизненного лечения; 3) требование высокой приверженности, трудновыполнимое для многих пациентов; 4) прогрессирование гиперактивации иммунной системы и ее последствий даже при условии высокой вирусологической эффективности лечения; 5) наличие побочных эффектов даже у современных лекарств; 6) формирование лекарственно-устойчивых вариантов вируса.
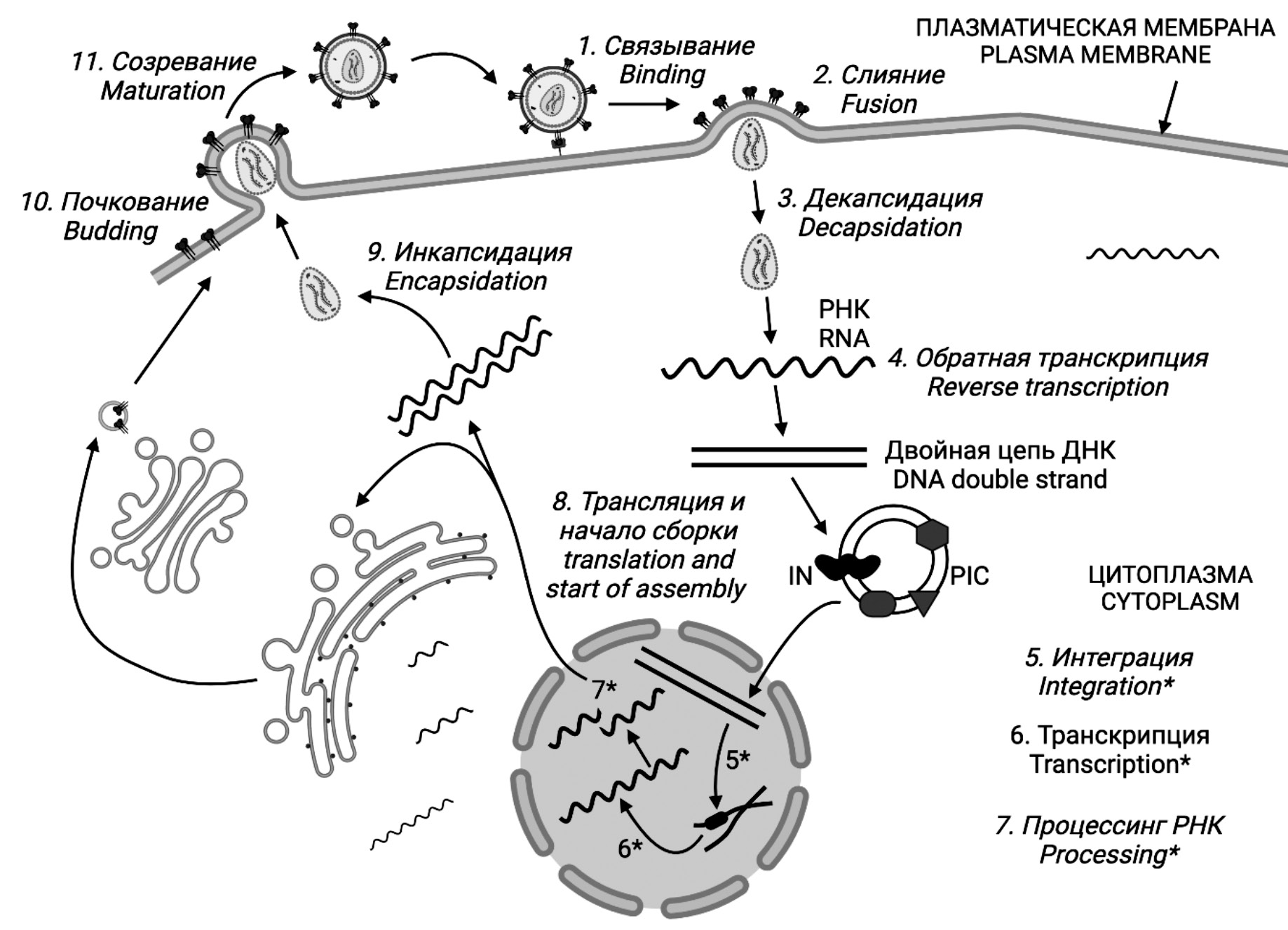
Последний из перечисленных феноменов связан с особенностью жизненного цикла ВИЧ, включающего стадию обратной транскрипции (рис. 1). Значительное число ошибок, совершаемых ферментом ВИЧ – обратной транскриптазой (reverse transcriptase, RT), затрагивает все области генома, включая позиции, определяющие связывание молекул лекарственных средств в составе мишеней АРТ. Результатом становятся мутации лекарственной устойчивости и снижение эффективности АРТ.

Рис. 1. Жизненный цикл ВИЧ. Рисунки 1‒10 созданы в программе BioRender (BioRender.com).
Fig. 1. HIV life cycle. Figures 1–10 were created in the BioRender program (BioRender.com).
Перечень мишеней АРТ ограничен и включает три фермента ВИЧ: RT, интегразу (integrase, IN) и протеазу (protease, PR), а также вирусные белки, участвующие в заражении клетки вирусом на этапах присоединения (связывания) и слияния (рис. 1). Феномен лекарственной устойчивости реализуется в отношении каждой из них, хотя и с разной частотой. На протяжении жизни примерно у 40% пациентов возникают устойчивые к АРТ штаммы ВИЧ, что требует замены схем лечения, как правило, на более дорогостоящие. Кроме того, устойчивые варианты ВИЧ могут передаваться в контакте и вызывать заражение, тем самым делая заведомо неэффективной уже первую схему АРТ. Инновационные препараты, направленные против тех же мишеней, хотя и реже, но вызывают формирование устойчивых вирусов и феномена в целом не отменяют.
Все это побуждает к поиску принципиально новых подходов к лечению ВИЧ-инфекции, в числе которых в последние годы все чаще обсуждается возможность разработки препаратов, направленных на взаимодействия ВИЧ с клеточными белками и структурами, важными для размножения вируса [1–7]. Ожидания от такого подхода заключаются в создании терапевтических альтернатив для лечения вирусных инфекций, не вызывающих лекарственной резистентности по причине того, что они имеют своей мишенью белки хозяина (host-targeted antivirals, HTA), генетически существенно более стабильные, чем вирусные.
Действительно, репликативные и биосинтетические механизмы клетки-хозяина играют критическую роль в жизненном цикле всех вирусов, являющихся по своей природе внутриклеточными паразитами. В этом кратком обзоре будет обсуждаться современное состояние исследований в этой сфере применительно к ВИЧ, включая провирусные и антивирусные клеточные факторы, функционирующие на разных этапах инфекционного цикла вируса, и некоторые подходы к разработке лекарственных препаратов, направленных на эти факторы.
Начало изобильному потоку публикаций на указанную тему было положено еще в 2008 г. сенсационной работой ученых из Гарварда [8], которые провели полногеномный скрининг генома человека с применением библиотеки из более чем 21 000 малых интерферирующих РНК (siRNAs) в поиске хозяйских факторов, участвующих в размножении ВИЧ. Последовательное «выключение» генов с последующей оценкой продукции р24-антигена в модельной системе, основанной на HeLa-производных клетках, позволило получить ошеломительный результат – как оказалось, 274 белка клеток человека так или иначе участвовали в репликации ВИЧ, причем до получения результатов этого эксперимента было известно только о 37 (13%) из них. Это участие осуществлялось на всех стадиях жизненного цикла ВИЧ – от присоединения вируса к клетке до почкования новых вирионов и проявлялось позитивной регуляцией продукции ВИЧ; за такими факторами закрепилось не вполне удачное название «факторы зависимости ВИЧ» (HIV-dependency factors, HDFs).
Строго говоря, поражало воображение число обнаруженных HDFs, но не факт их существования, ведь очевидно, что обладающий столь минимальным набором собственных возможностей вирус непременно должен прибегать к помощи белков-посредников для реализации всех этапов размножения. Последующие исследования показали, что речь идет не просто о приспособлении процессов репликации вируса в клетке к существующей для нужд самой клетки машинерии, но и об активном вмешательстве ВИЧ в широкий спектр клеточных процессов, которые включают ремоделирование эндомембран, полимеризацию и организацию цитоскелета, модуляцию экспрессии генов и белков-хозяев, апоптоз и деление клеток, уклонение от иммунного ответа и многое другое [2, 6, 7, 9]. Далеко не все HDFs детально изучены, однако о некоторых из них уже вполне можно говорить как о потенциальных мишенях терапии.
Еще более увлекательной становилась картина по мере того, как накапливались данные о существовании другой группы белков, пусть и не столь многочисленной, зато очень перспективной с точки зрения разработки терапевтических средств. Речь идет о так называемых факторах рестрикции (restriction factors, RFs) – белках человека, способных блокировать размножение ВИЧ в клетках. Механизмы, с помощью которых они это делают, чрезвычайно затейливы, отличаются большим разнообразием и являются сейчас предметом самого активного изучения.
Некоторые примеры белков, относящихся к обеим группам – «пособников» и «противников» ВИЧ, будут приведены далее в порядке, приблизительно соответствующем порядку событий размножения вируса в клетке. Цикл репликации ВИЧ включает следующие этапы: 1) присоединение вирусных частиц и последующее 2) слияние клеточной и вирусной мембран с участием рецепторов клетки; 3) декапсидация («раздевание») вирусных частиц; 4) обратная транскрипция с участием обратной транскриптазы ВИЧ и образованием комплементарной ДНК (кДНК); 5) формирование преинтеграционного комплекса (preintegration complex, PIC) и интеграция кДНК в хроматин клетки с участием интегразы ВИЧ; 6) транскрипция с участием клеточной РНК-полимеразы; 7) процессинг (сплайсинг) РНК и экспорт ее в цитоплазму; 8) синтез белков и сборка новых вирусных частиц; 9) инкапсидация с образованием внутренней структуры частицы; 10) отпочковывание частиц от клетки с присоединением оболочечных белков; 11) созревание с участием протеазы ВИЧ (рис. 1).
В составе вириона, заражающего чувствительную клетку, всегда, помимо структурных белков, присутствует небольшое количество ферментов – RT, IN и PR, а также некоторые клеточные белки, захваченные вирионом в момент отпочковывания от клетки и необходимые на ранних этапах размножения вируса.
Присоединение. Первыми клеточными белками, с которыми встречается ВИЧ, являются клеточные рецепторы (CD4 и CCR5), необходимые для присоединения вируса. Присутствие первого из них на мембране клетки абсолютно необходимо для ее заражения, отсутствие второго корецептора (CCR5) в случаях гомозиготного дефекта – делеции CCR5D32 – в подавляющем числе случаев защищает от заражения, однако оставляет редкую возможность инфицирования вирусами, тропными к альтернативному корецептору CXCR4. Подробно этот феномен был многократно описан в литературе (рис. 2) [10–12].

Рис. 2. Этапы проникновения ВИЧ в клетку.
Fig. 2. Stages of HIV entry into the cell.
Препарат, направленный на ингибирование корецептора CCR5, уже существует и применяется под названием «Маравирок» (MVC); новые ингибиторы CCR5 и CXCR4 находятся на стадии клинических испытаний; ингибиторы CD4 также находятся в разработке и вместе с ингибиторами корецепторов формируют класс антагонистов рецепторов ВИЧ.
Помимо хорошо известных поверхностных рецепторов CD4 и CCR5, идентифицированы еще несколько дополнительных факторов, функция которых критична для заражения клеток ВИЧ.
Известно, что ВИЧ способен заражать клетки не только путем взаимодействия «вирус–клетка», но и в результате прямого контакта клеток, не требующего наличия рецепторов. Такой способ заражения («клетка–клетка») многократно превосходит по эффективности «классический» вариант и находит поддержку в лице молекул ALCAM (activated leukocyte cell adhesion molecule) – гликопротеинов из суперсемейства иммуноглобулинов, обладающих сильно выраженными адгезивными свойствами. ALCAM опосредуют межклеточную адгезию и способствуют эффективному распространению вируса посредством прямой передачи от клетки к клетке (рис. 2) [13, 14].
Еще одна молекула с адгезивными свойствами – DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin). На поверхности основных мишеней ВИЧ – CD4+-Т-клеток – ее нет, но зато много на макрофагах и особенно дендритных клетках, первыми встречающих вирус на периферии при заражении. DC-SIGN – лектин С-типа, который распознает гликопротеины на поверхности микроорганизмов и в случае ВИЧ-инфекции выступает в роли неспецифического корецептора, взаимодействующего с gp120. Последующая интернализация комплекса DC-SIGN–gp120 приводит к формированию внутриклеточных «хранилищ» вируса, и после рециркуляции комплекса на поверхность клетки инфицирование CD4+-Т-клеток в лимфоидных органах вследствие прибытия в них дендритных клеток существенно облегчается [15].
Для выполнения функции корецептора CCR5 очень важна сульфатная группа в составе его N-концевого тирозина. В сульфатировании этой аминокислоты участвуют сразу два клеточных белка – SLC35B2 (solute carrier family 35 member B2) и TPST2 (tyrosylprotein sulfotransferase 2) (рис. 2). В роли донора активированного сульфата выступает PAPS (3’-phosphoadenosine 5’-phosphosulfate transporter), который синтезируется в цитозоле и при помощи SLC35B2 перемещается в просвет аппарата Гольджи, где резидентный белок TPST2 катализирует сульфатирование тирозина CCR5. Как оказалось [14], отсутствие TPST2 и/или SLC35B2 полностью предотвращает заражение клеток ВИЧ.
Всем описанным выше белкам клетки, способствующим заражению ВИЧ, противостоит фактор рестрикции SERINС5 (serine incorporator 5), обнаруживаемый в составе вирусных частиц. Его природная функция мало изучена; известно, что этот белок имеет трансмембранный домен из 10 звеньев и является, таким образом, родственным корецептору CCR5 [16]. Механизм его участия в репликации ВИЧ также не вполне ясен; есть предположения о том, что он способен связывать белок gp120 и тем самым препятствовать процессу слияния мембран, при этом gp120 «задерживается» на поверхности мембраны и становится объектом действия антител к ВИЧ (рис. 2) [4, 17, 18].
Антагонистом SERINС5 является вирусный белок Nef, который способен удалять его из состава мембраны и секвестрировать в эндосому с последующей деградацией [19].
Декапсидация. До недавнего времени считалось, что после попадания ретровируса в цитозоль клетки-хозяина происходит немедленное сбрасывание капсида с высвобождением РНК и последующей обратной транскрипцией. Этой точке зрения в наши дни противостоит другая, справедливо обращающая внимание на то, что кДНК, образующаяся в ходе обратной транскрипции, может сделаться объектом молекулярных сенсоров [20, 21], чувствительных к присутствию ДНК в цитозоле, и вызвать ненужную для вируса иммунную реакцию. Избежать этого можно, если защитить образующуюся кДНК белковой оболочкой, именно поэтому теперь многие исследователи считают, что процесс декапсидации и обратная транскрипция сопряжены во времени, при этом транскрипция начинается в составе нуклеокапсида, а сбрасывание капсида происходит по мере продвижения кДНК к ядру и заканчивается непосредственно перед импортом PIC (см. далее) в ядро [3, 22] либо даже в ядре (рис. 3) [23].

Рис. 3. Факторы зависимости (CypA) и рестрикции (TRIM5α) на этапе декапсидации ВИЧ.
Fig. 3. Dependency (CypA) and restriction (TRIM5α) factors at the HIV decapsidation stage.
Центральным звеном всех взаимодействий на этом этапе оказывается вирусный капсидный белок (СА) p24, соcтавляющий внутреннюю оболочку вириона. За право контактировать с ним борются представители обеих «группировок» клеточных белков, при этом наиболее известными из них являются RF TRIM5α (tripartite motif-containing protein 5) и циклофилин из группы HDFs. Механизм функционирования обоих изучен пока недостаточно, и далее изложены лишь некоторые обоснованные предположения о событиях с их участием.
TRIM5α непосредственно связывает молекулы CA нуклеокапсида вирусов, вновь проникших в цитоплазму после завершения слияния вирусной и клеточной мембран. Димеризация СА нарушает архитектуру капсида и вызывает его ускоренную фрагментацию, тем самым отменяя обратную транскрипцию (рис. 3) [3, 24]. Белок TRIM5α есть у всех приматов, однако описанная выше функция его видоспецифична: TRIM5α от обезьян Старого Света, таких как макаки резус, ограничивают широкий спектр ретровирусов, включая ВИЧ, а человеческий TRIM5α эффективно препятствовать ВИЧ не способен [3], хотя есть сведения о полиморфных вариантах TRIM5α, ассоциированных с замедленной прогрессией ВИЧ-инфекции [25].
Возможно, причина недостаточного эффекта TRIM5α состоит в наличии в клетках человека эффективно работающего циклофилина (сyclophilin A, CypA). Этот белок из группы шаперонов присутствует в клетке в значительных количествах и формах, принимая участие в процессах укладки (folding) белковых молекул. Его можно встретить на разных этапах репликации ВИЧ, и в составе вирусных частиц он тоже всегда есть, видимо для того, чтобы начать свою деятельность сразу после заражения клетки. Предполагается, что механизм повышения инфекционности ВИЧ в присутствии CypA объясняется его способностью взаимодействовать с CA, при этом стабильность капсида повышается, что, в свою очередь, позволяет сохранить в целости кДНК до момента пересечения ею ядерной мембраны [5, 22, 23]. Простое объяснение этого факта заключается в конкуренции CypA с TRIM5α за связывание с СА, чуть более сложное, но и вполне вероятное, – в непосредственном взаимодействии TRIM5α–CypA, создающем стерическое препятствие для его контакта с СА (рис. 3) [26]. Замысел будущих разработок лекарственных АРТ-препаратов включает создание неиммуносупрессивных ингибиторов CypA, а также повышение эффективности связывания TRIM5α–СА [6, 26].
Цитоскелет клетки. Размеры вирусных и субвирусных компонентов поступающих в клетку вирионов не позволяют им свободно диффундировать в «густонаселенной» цитоплазме, поэтому ранние этапы жизненного цикла ВИЧ критически зависят от процессов цитоплазматического транспорта.
Один из белков клетки, вовлеченных в организацию этого процесса, – сократительный белок актин (actin). Его микрофиламенты выполняют свою функцию на периферии клетки и, возможно, связаны с процессом эндоцитоза вирусных частиц (см. выше) [6].
Роль микротрубочек неоднократно обсуждалась в отношении как минимум двух аспектов жизненного цикла ВИЧ – слияния мембран и внутриклеточного перемещения поступающих капсидов. Белками клетки, ассоциируемыми с трафиком, являются MAP1A и MAP1S (microtubule-associated proteins), а также динеин (dynein), составляющие основную часть микротрубочек. О механизмах их функционирования известно лишь то, что они способны связывать вездесущий CA в составе всех вирусных компонентов и направлять их нужным курсом, то есть в сторону клеточного ядра [6, 23].
Поскольку элементы цитоплазматического трафика составляют значительную часть белков клетки хозяина и необходимы для всех внутриклеточных процессов, представить их себе в роли терапевтических мишеней (например, путем деполимеризации) довольно трудно. С учетом того, что микрофиламенты и микротрубочки физически и функционально переплетаются, взаимодействие между ВИЧ и цитоскелетом представляется еще более сложным и требует продолжения исследований.
Обратная транскрипция. На этом важнейшем этапе размножения ВИЧ наибольшую известность приобрели два RFs – APOBEC3G и SAMHD1.
APOBEC3G (apolipoprotein B mRNA editing enzyme, catalytic subunit 3G) в ходе синтеза кДНК функционирует как дезаминаза, превращая дезоксицитидин (dC) в дезоксиуридин (dU) в составе минус-цепи. На следующем этапе результирующая плюс-цепь ДНК содержит множество гипермутаций G-A, приводящих к формированию преждевременных стоп-кодонов и продукции аберрантных вирусных транскриптов, которые со временем подвергаются деградации (рис. 4).

Рис. 4. Факторы рестрикции APOBEC3G и SAMHD1 на этапе обратной транскрипции ВИЧ.
Fig. 4. The restriction factors APOBEC3G and SAMHD1 at the HIV reverse transcription stage.
У белка APOBEC3G есть свой антагонист среди белков ВИЧ – Vif, также обнаруживаемый в составе вирионов. Связываясь с APOBEC3G, Vif опосредует его протеосомную деградацию с использованием механизма убиквитинирования [4, 19, 27].
Клеточный белок SAMHD1 (sterile a motif domain-, HD domain-containing protein 1), действуя в роли фосфогидролазы, осуществляет превращение нуклеотид-трифосфатов (dNTPs) в дезоксинуклеотиды, тем самым истощая ресурс для синтеза кДНК (рис. 4). В дополнение к этому, результирующее снижение уровня ДНК в цитозоле ограждает клетку от включения молекулярных сенсоров и последующей нежелательной секреции интерферона и хронического воспаления [3, 6, 23]. У ВИЧ-1 нет белка – антагониста SAMHD1, а у ВИЧ-2 есть – это белок Vpx, который участвует в деградации SAMHD1 [3, 27].
Формирование преинтеграционного комплекса. После завершения синтеза молекул кДНК им предстоит встроиться в хромосомную ДНК клетки. В отличие от прочих ретровирусов, которые обычно заражают клетки в стадии митоза, когда ядерная мембрана отсутствует и ДНК легкодоступна, ВИЧ обладает способностью заражать неделящиеся клетки, то есть клетки, находящиеся в периоде интерфазы. Поскольку продолжительность этой фазы существенно превышает длительность митоза, такая возможность предоставляет вирусу колоссальные преимущества, обеспечивая ему огромное число мишеней и высочайший уровень репликации. В неделящихся клетках ядро окружено плотной оболочкой – ядерной мембраной, поэтому пассивной диффузией вопрос не решается и требует активного транспорта кДНК. Преинтеграционный этап и собственно интеграция являются центральным и наиболее сложно организованным событием размножения ВИЧ.
Преинтеграция начинается с формирования минимальной структуры – интасомы, состоящей из кДНК и фермента IN; вначале IN имеет димерную структуру, а на этапе интеграции в составе функционального PIC находится в форме тетрамера [28]. Молекулы IN «сводят» между собой концы кДНК, после чего образуется незамкнутое кольцо, на концах которого находятся длинные концевые повторы (LTR). После присоединения нескольких коровых вирусных и клеточных белков образуется PIC (рис. 5).

Рис. 5. Участие нуклеопоринов в транслокации преинтеграционного комплекса ВИЧ.
Fig. 5. Involvement of nucleoporins in the translocation of the HIV pre-integration complex.
В составе PIC в цитоплазме происходит первый этап интеграции ВИЧ – 3’-процессинг кДНК, в котором на первый план выходит деятельность IN. В ходе 3’-процессинга на каждом из 3’-концов вирусной ДНК в составе PIC удаляются по два нуклеотида с образованием свободных гидроксильных (ОН)-групп, и в ядро кДНК попадет уже в процессированном виде. На этом этапе возникает проблема, требующая «помощи» со стороны клетки. Заключается она в том, что в окружении PIC находится заметное число линейных молекул кДНК, которые с помощью IN легко могут «самоинтегрироваться» и тем самым снизить эффективность последующей истинной интеграции. Предотвращение этого феномена происходит при содействии клеточного фактора BAF (barrier-to-autointegration factor) – ДНК-связывающего белка, способного конденсировать ДНК [29]. Защиту от подобных суицидальных событий также обеспечивают несколько ДНКаз, связанных с эндоплазматическим ретикулумом [30].
Транслокация преинтеграционного комплекса. После завершения 3’-процессинга PIC начинает двигаться по направлению к ядру клетки. Транслокация PIC обеспечивается взаимодействием кариофильных вирусных белков, содержащих особые последовательности аминокислот – сигналы ядерной локализации (nuclear localization signal, NLS), с клеточными белками, функционирующими на всех этапах ядерного транспорта. К числу вирусных белков, имеющих NLS, относятся MA, IN, а также неструктурный белок ВИЧ Vpr [31].
В составе ядерной мембраны есть поры (nuclear pore complex, NPC), в основном состоящие из высоко консервативных нуклеопоринов (nucleoporins, NUPs) [32]. Они обеспечивают транспорт крупных гидрофильных молекул с массой более 40 kDa, при этом все остальные молекулы проникают в ядро путем пассивной диффузии. В зависимости от выполняемой функции NUPs могут располагаться в цитозоле либо в нуклеоплазме.
Около 30 белков этой группы образуют NPC, имеющий кольцевую структуру: цитоплазматическое кольцо с 50 филаментами (примером может быть Nup358), ядерное кольцо («корзинка» из 8 филаментов), включающее Nup153, и туннель между ними (Nup170, Nup155 и пр.) [32]. Внутренние размеры пор значительно меньше (около 8 нм), чем диаметр PIC (~ 28 нм) [33], поэтому PIC приходится в буквальном смысле «протискиваться» через каналы в мембране, при этом энергетические затраты обеспечивает аденозинтрифосфат (АТФ).
Для транслокации PIC может непосредственно контактировать с компонентами NPC либо использовать растворимые белки-транспортеры из группы кариоферинов [22]. Природная функция этих белков состоит в транспортировке факторов сплайсинга мРНК в ядро клетки; в клетке, инфицированной ВИЧ, наиболее известными «помощниками» ВИЧ являются кариоферины TNPO3 (transportin-3) и CPSF6 (cleavage and polyadenylation specificity factor 6). Непосредственно с CA, не имеющим собственного NLS, взаимодействует, по-видимому, лишь CPSF6, а задача TNPO3 состоит в том, чтобы осуществлять нарезание и полиаденилирование CPSF6. В качестве альтернативы этим белкам может выступать Nup358, также имеющий сродство к CA [34]. Наконец, в процессе интеграции активно участвует все тот же CypA, закрепляя PIC на цитоплазматической стороне ядерной мембраны и направляя его в ядерную пору; считается [22, 31], что в этом как-то участвует и вирусный белок Vpr (рис. 5).
Прохождение через поры обеспечивается несколькими NUPs, каждый из которых взаимодействует с одним или более компонентами PIC. На ядерной стороне мембраны PIC встречает белок NUP153, который локализуется в конце «туннеля» NPC и непосредственно связывает IN, CA и Vpr ВИЧ-1.
Отметим здесь, что процесс транслокации PIC включает большое число участников как со стороны вируса (здесь перечислены не все даже из хорошо изученных), так и со стороны клетки, при этом функции каждого из них дублируются, и на каждом этапе имеется выбор в пользу того или иного компонента в зависимости от состояния процесса размножения вируса.
Все перечисленные выше факторы, в конечном счете способствующие репликации ВИЧ-1, можно отнести к группе HDFs. На данный момент описан только один внутриклеточный RF, ингибирующий транслокацию PIC, – это белок MX2 (от Myxovirus resistance, он же GTPase MxB). Индуцируемый интерфероном MX2 имеет сродство к CA и концентрируется на цитоплазматической стороне ядерной мембраны [35]. О механизмах его действия известно мало; MX2 ингибирует разные штаммы ВИЧ-1, а также другие лентивирусы приматов, но минимально активен в отношении лентивирусов, не относящихся к приматам. Некоторые штаммы ВИЧ-1 проявляют естественную устойчивость к MX2 без каких-либо очевидных последствий для фитнеса вируса [36].
Собственно интеграция ВИЧ. Попав внутрь ядра, PICs ВИЧ-1 начинают медленное диффузное движение и преимущественно концентрируются на периферии ядра [37], не слишком удаляясь от ядерных пор. Последовательность хромосомной ДНК в участке встраивания провируса не имеет существенного значения, однако назвать локализацию этого процесса полностью случайной было бы неправильно. В выборе участка интеграции ведущую роль играет, по-видимому, доступность ДНК, то есть окружающая ее структура хроматина. В неделящихся клетках значительная часть ДНК клетки находится в конденсированном состоянии (гетерохроматин) и связана с гистонами, поэтому ВИЧ-1 преимущественно связывается с деконденсированным хроматином (эухроматином), а «горячими точками» для интеграции являются транскрипционно активные участки ДНК, освобожденные от гистонов [38]. По всей видимости, это не просто облегчает процесс встраивания, но и дает вирусу эволюционные преимущества, обеспечивая ему в последующем высокий уровень транскрипции в тандеме с геном-мишенью [6].
Интересно, что клеточные гены, которые чаще других становятся объектами встраивания провируса ВИЧ-1, так называемые «гены рекуррентной интеграции», также концентрируются на периферии ядра и контактируют с ядерными порами. Организованный таким образом процесс позволяет свести затраты времени на собственно интеграцию кДНК к минимуму.
Этот процесс в значительной степени регулируется капсидом и белками, взаимодействующими с СА [22, 39], что вновь указывает на хорошую координацию проникновения кДНК в ядро и интеграции. Выбор локуса интеграции может быть организован иерархически: топологическая локализация определяется компонентом NPC NUP153, тогда как доминирующая роль в выборе участка транскрипционно активного хроматина и последующего встраивания принадлежит клеточным белкам CPSF6, LEDGF/p75 (lens epithelium-derived growth factor) и INI1 (INtegrase interactor protein 1) [37, 40], а также, разумеется, и самой IN ВИЧ-1 (рис. 6).

Рис. 6. Интеграция провирусной ДНК ВИЧ.
Fig. 6. Integration of HIV proviral DNA.
Считается, что CPSF6, в ассоциации с CA на этапе транслокации обеспечивший проникновение PIC в ядро, играет решающую роль и в локализации PIC в ядре, направляя его в эухроматин [40]. Что касается INI1, то, по некоторым предположениям [41], этот фактор может стабилизировать PIC в клетке-хозяине, поддерживая IN в устойчивой конформации, которая предотвращает неспецифические взаимодействия и самоинтеграцию. Белок BAF, ответственный за профилактику самоинтеграции в составе PIC [29], продолжает выполнять эту функцию и на этапе интеграции [28, 31, 39].
Белок LEDGF/p75, изученный лучше других, служит связующим звеном (или мостом) между преинтеграционным комплексом и ДНК хозяина. Последовательность связывания LEDGF/p75 с ДНК хозяина и PIC остается неясной, однако независимо от последовательности событий считается, что присутствие LEDGF/p75 приводит к сближению димеров IN друг с другом с образованием тетрамера. Этот аллостерический эффект приводит к активации IN, которая после этого переходит к выполнению своей основной функции – реакции переноса цепи (strand transfer) [29, 42]. Она заключается в том, что c использованием ранее сформированных гидроксильных групп IN разрезает хромосомную ДНК в выбранном участке и одновременно соединяет ее 5’-фосфатные группы с концами вирусной кДНК. Промежуточным результатом становится участок ДНК, имеющий разрывы и свободные 5’-концы вирусной ДНК; этот временный дефект восстанавливается инструментами клетки-хозяина с образованием интегрированного провируса [29, 42].
Интерес к LEDGF/p75 возник более десятка лет назад [43] и реализовался в разработке группы препаратов, мишенью которых является интерфейс между активным центром IN и связывающим его доменом LEDGF (LEDGINs). Действие этих ингибиторов основано на аллостерическом эффекте и проявляется двумя феноменами: ранним, то есть очевидным снижением активности IN и интеграции провируса, и поздним, который приводит к морфологическим изменениям продуцируемых вирусных частиц и нарушениям архитектуры кора вируса вследствие повышенной мультимеризации IN [43–46], при этом рибонуклеопротеин оказывается за пределами кора. Такие «увечные» частицы составляют до 70% и не способны во время последующего раунда заражения обеспечить нормальный ход событий размножения вируса [46]. В доклинических испытаниях был подтвержден также негативный эффект некоторых LEDGINs в отношении формирования вирусных резервуаров [47], и это дает основания для надежд на применение этих препаратов в целях функционального излечения ВИЧ-инфекции [47, 48] уже в ближайшем будущем.
Транскрипция. Собственных инструментов для транскрипции провирусной ДНК у ВИЧ нет, и для транскрипции своего генома вирус привлекает все необходимые компоненты клеточного аппарата, и прежде всего РНК-полимеразу. Для того, чтобы их «завербовать» в пользу работы на вирус, используется главнейший из регуляторных факторов ВИЧ – белок Tat. Именно от его активности зависит уровень транскрипции ВИЧ, а отсутствие Tat приводит провирусную ДНК в состояние латентности. События, которые при этом происходят, многократно описывались в литературе [49–52] и здесь подробно рассматриваться не будут.
Кратко, сигналом для инициации считывания РНК становится контакт между активаторными белками SP1 (specificity protein 1) и NF-κB (nuclear factor [kappa]B), каждый из которых располагается в соответствующем участке промотора LTR. После этого РНК-полимераза (RNA-PII), распознав TATA-box, приступает к синтезу РНК, однако в отсутствие Tat синтез быстро останавливается, и результатом этого этапа транскрипции становится короткий фрагмент РНК, образующий петлю TAR (рис. 7).

Рис. 7. Активность хозяйских факторов на этапе транскрипции ВИЧ.
Fig. 7. Activity of host factors at the HIV transcription stage.
C появлением Tat ситуация кардинальным образом меняется, поскольку он инспирирует формирование комплекса P-TEFb (positive transcription elongation factor b), включающего CDK9 (сyclin-dependent kinase 9) и CycT (Cyclin T). Взаимодействие TAR/Tat/P-TEFb с «апатичной» RNA-PII вызывает ряд ее модификаций (главным образом фосфорилирование) и приводит в активное состояние, после чего полимераза переходит к продуктивной элонгации с образованием полноразмерных мРНК ВИЧ.
Все указанные выше (а также неуказанные) клеточные факторы транскрипции способствуют формированию мРНК ВИЧ и по определению являются, таким образом, HDFs. Судьба образующихся мРНК, однако, складывается не всегда благополучно, поскольку на этапе транскрипции обнаружены RFs, способные приводить к деградации РНК.
Одним из них является РНКаза-L – медиатор противовирусной активности, индуцированный интерфероном-1. Этот фермент обладает широким спектром активностей, связанных с регуляцией рибосомальных и вирусных РНК и, в частности, способен проявлять рибонуклеазные свойства [53]. Эффективная деградация мРНК ВИЧ с участием РНКазы-L может препятствовать вхождению ВИЧ в продуктивный цикл размножения и способствовать поддержанию его латентного состояния.
Еще один фактор, вызывающий деградацию мРНК ВИЧ, – ZAP (zinc finger antiviral protein). Этот белок обладает широкой противовирусной активностью и ограничивает не только ретровирусы, но и многие другие РНК- и ДНК-вирусы. Механизм его функционирования долго оставался неясным, и лишь недавно стало понятно, что мишенью ZAP являются динуклеотиды CpG в вирусной РНК, за распознаванием которых следует рекрутирование компонентов экзосомного комплекса для деградации РНК (рис. 7) [53].
Другой механизм рестрикции использует белок KAP1 (Kruppel-associated box (KRAB)-interacting protein 1), который ослабляет взаимодействие RNA-PII с комплексом P-TEFb и задерживает ее таким образом в составе промотора, снижая эффективность элонгации [54].
Ядерный экспорт. По завершении транскрипции образуется лишь один вид молекул РНК, имеющих размеры, соответствующие размерам провируса. Как оказалось, судьба вновь синтезированных полноразмерных молекул РНК не предопределена, и одни и те же молекулы могут выступать в роли матричной либо геномной РНК, встраиваясь в будущие вирусные частицы в ходе сборки [2, 51]. Существование таких несплайсированных транскриптов ВИЧ-1 противоречит клеточному «внутреннему распорядку», поэтому для их «спасения» разработана специальная стратегия с участием вирусного белка Rev и клеточных «помощников».
По завершении транскрипции полноразмерный транскрипт мРНК ВИЧ-1 вначале подвергается сплайсингу с образованием мРНК белков ВИЧ-1 Rev, Tat и Nef [51], после чего мРНК Rev транспортируется в цитоплазму для трансляции Rev. Белок Rev, имеющий NLS, возвращается в ядро с участием импортинов. На поздних стадиях размножения ВИЧ-1, когда достигается достаточная концентрация Rev, он приступает к выполнению своей основной функции по выводу несплайсированных мРНК из ядра.
В составе Rev есть сигнал ядерного экспорта (nucleus export signal, NES), с помощью которого он связывает белок из группы кариоферинов CRM1 (chromosome maintenance region 1), известный также под именем экспортина-1, и выводит полноразмерные транскрипты в цитоплазму, где они становятся матрицей для трансляции структурных белков Gag и Pol, а также входят в состав новообразованных вирионов. Прочие продукты сплайсинга необходимы для формирования вспомогательных белков Vif и Vpr, а также оболочечных белков Env [23]. Для осуществления собственно сплайсинга используются те же рибонуклеиновые комплексы (spliceosomes), которые применяет клетка для выполнения этой задачи в отношении собственных белков, при этом имеются сведения о том, что белок Rev вовлекает в это событие клеточные РНК-хеликазы [55], выступающие в роли молекулярных «шаттлов» для коротких транскриптов ВИЧ (рис. 8).

Рис. 8. Сплайсинг и ядерный экспорт мРНК ВИЧ.
Fig. 8. Splicing and nuclear export of HIV mRNAs.
Ядерный экспорт мРНК, как и импорт PIC, требует прохождения через ядерные поры (NPC) и опосредуется растворимыми рецепторами-шаттлами, которые перемещаются между ядром и цитоплазмой. Помимо описанного выше CRM1, на этапе экспорта функционирует белок TAP-p15 [32], а также около десятка менее изученных клеточных факторов, которые также присоединяются к мРНК с образованием mRNP (messenger ribonucleoprotein). Дальнейшие события развиваются следующим образом: mRNP взаимодействует с NUPs, инициируя экспорт. После успешной «стыковки» mRNP продвигается по внутреннему каналу NPC, контактируя с NUPs, и высвобождает мРНК с участием цитоплазматических филаментов NPC NUP214 [32].
Трансляция и сборка вирионов. На этом этапе размножения, как и на предыдущем, ВИЧ полностью зависим от клеточного аппарата биосинтеза, однако, применяя множество ухищрений, эффективно изменяет соотношение продукции вирусных и клеточных белков в свою пользу. Среди приспособлений, которые использует ВИЧ для повышения уровня и регуляции синтеза собственных белков, находятся использование энхансеров трансляции – дополнительного участка посадки рибосомы IRES (internal ribosome entry site) и PCE (post-transcriptional control element), усиливающего связь с полисомой; механизм leaky scanning для прочтения двух белков с общей рамкой считывания; рибосомное сканирование (ribosome scanning) и пр. Самым известным из «изобретений» ВИЧ является феномен «игнорирования» стоп-кодона и сдвига рамки считывания на уровне трансляции, позволяющий регулировать соотношение продукции структурных белков и ферментов, кодируемых одним и тем же геном pol.
Эти механизмы, будучи крайне интересными, не имеют прямого отношения к теме настоящего обзора и подробно освещены в ряде работ [2, 19, 23, 51, 56], а здесь будут приведены примеры менее известных случаев взаимодействия ВИЧ c хозяйскими факторами на этапе трансляции и последующей сборки.
После окончания процесса сплайсинга и экспорта всех РНК в цитоплазму наступает этап инициации трансляции, определяющий скорость размножения ВИЧ в целом. Инициирующие элементы есть в каждом из транскриптов ВИЧ, что позволяет им транслироваться независимо друг от друга и само по себе способствует повышению уровня продукции вируса. Белки Gag, Pol, Tat и Rev синтезируются отдельно от высоко модифицированных гликопротеинов Env, встречаясь с ними уже на этапе почкования (рис. 9).

Рис. 9. Факторы хозяйской клетки на этапе трансляции ВИЧ.
Fig. 9. Host cell factors at the HIV translation stage.
Эффективность инициации определяется частотой «посадки» мРНК на полирибосому (полисому, polysome) и прочностью связывания с ней. На эти процессы влияют сразу несколько белков – одни способны их усиливать, например регулятор сплайсинга 9G8, белок Sam68 (Src-associated in mitosis 68), РНК-хеликаза А (RHA), а другие, напротив, играют роль факторов рестрикции, ингибируя или отменяя события инициации. К ним относится белок IFITM (interferon-induced transmembrane protein), способный исключать взаимодействие мРНК с полисомой [23]; его вирусным антагонистом является белок Nef. Другой пример – hnRNP E1 (heterogeneous nuclear ribonucleoprotein E1), который обладает способностью разобщать большую и малую субъединицы рибосомы [2].
По мере приближения финального этапа размножения – сборки (assembly) и почкования (budding) вирионов – соперничество двух групп белков проявляется все сильнее. После достижения достаточной концентрации продуктов биосинтеза – вирусных белков – начинается процесс сборки вирионов, при этом главная роль в организации внутренней структуры частиц, включения РНК и продвижения к мембране клетки принадлежит вирусным белкам группы Gag [56]. На начальном этапе сборки вирусные частицы являются незрелыми, и лишь после нарезания предшественника Gag-Pol формируются все внутренние белки, в частности СА, формирующий капсидную часть вириона конической формы.
Как выяснилось [57], на последующих этапах ВИЧ активно использует для почкования комплекс клеточных белков аппарата ESCRT (endosomal sorting complexes required for transport), участвующий в процессах ремоделирования мембран. Известный своей активной деятельностью белок СА, взаимодействуя с компонентом ESCRT – белком TSG101 (tumor susceptibility gene 101), способствует перемещению формирующихся вирусных частиц в эндосомы с последующим образованием мультивезикулярных телец (multivesicular body, MVB) (рис. 10). В таком виде будущие вирионы могут сохраняться в цитоплазме вплоть до момента почкования.

Рис. 10. Сборка и почкование вирусных частиц ВИЧ.
Fig. 10. Assembly and budding of HIV viral particles.
Белок Env синтезируется в виде предшественника gp160 в шероховатом ретикулуме (EPR) и подвергается созреванию/гликозилированию с образованием gp120 и gp41 с участием аппарата Гольджи. Перемещению из EPR к месту окончательного формирования ему помогает клеточный белок Rab11-FIP1C (FIP1C) (Rab coupling protein) [56]. Предпочтительными местом последующего «заякоривания» Env в мембране становятся «липидные плотики» (lipid rafts), обогащенные холестерином и насыщенными жирными кислотами.
Кульминация наступает на этапе собственно почкования, когда проявляется эффект сразу нескольких факторов рестрикции. Два из них – белки GBP5 (guanylate binding protein 5) и MARCH8 (E3 ubiquitin ligase membrane-associated RING-CH 8) – препятствуют «нарезанию» gp160 и тем самым понижают эффективность включения белков Env в новые вирионы; результатом становится формирование неинфекционных вирусных частиц. В дополнение к этому MARCH8 обладает способностью удерживать Env в цитоплазме с помощью не вполне ясного механизма. Белок PSGL-1 (P-selectin glycoprotein ligand 1) ограничивает динамику актина вблизи клеточной мембраны и задерживает Env в ее составе [56].
Самым изученным из факторов рестрикции, действующих на этапе сборки, является тетерин (tetherin, BST-2, bone marrow stromal antigen 2). Этот белок имеет необычную структуру, включающую Т-концевой трансмембранный и С-концевой гликозилированный концы, что позволяет ему удерживаться в мембране и одновременно захватывать вновь образованные вирионы. BST-2 связывает между собой липидные «плотики» и молекулы актина, тем самым непосредственно ограничивая подвижность вирионов в тот момент, когда они уже готовы отделиться от мембраны [3, 16, 19, 56] (рис. 10). Тетерин существенно снижает заражение по типу «вирус–клетка» и в меньшей степени – по типу «клетка–клетка» [16]; на этом основана точка зрения, в соответствии с которой заражение по типу «вирус–клетка» преимущественно происходит при передаче вируса, в то время как на стадии его распространения в организме путь «клетка–клетка» преобладает.
В роли антагониста BST-2 выступает вирусный белок Vpu, который связывает N-концевую область цитоплазматического домена BST-2, удаляет его из мембраны и стимулирует его деградацию по протеасомальному или лизосомальному пути; альтернативный вариант – простое вытеснение BST-2 из участка сборки вирионов [58]. Интересно, что у ВИЧ-2 нет белка Vpu, и его функцию берет на себя белок Nef [59].
Этот последний пример наводит на размышления, касающиеся совместной эволюции ВИЧ и его природных хозяев, наблюдающейся на протяжении многих десятков тысяч лет. Факторы рестрикции – клеточные белки, каждый из которых имеет функцию, связанную с жизнеобеспечением клетки. Нередко выполнение этой функции «пересекается» с репликативным циклом вируса и проявляется мощной ингибирующей активностью. Это обстоятельство поставило перед вирусом задачу приспособления, с которой он изобретательно справился, в ходе эволюции приобретя дополнительные белки, к тому же обладающие возможностью перекрывания и «подстраховки» обязанностей. Понимание структуры и функции этих вспомогательных белков и механизмов их взаимодействия с лигандами в клетке-хозяине может иметь важное значение для разработки новых методов лечения ВИЧ-инфекции.
Еще один аспект, касающийся будущих разработок, связан с детальным изучением природных функций самих клеточных факторов – как HDFs, так и RFs. Первые из них предположительно должны стать объектом ингибирования, а вторые быть стимулированы с целью ограничения размножения ВИЧ.
Разработка препаратов с ингибирующими свойствами – традиционная задача фармакологии, однако ингибирование клеточных белков с разнообразными функциями потенциально всегда таит в себе опасность нарушения метаболизма клетки и последующей за ним токсичности, поэтому вполне очевидно, что из множества HDFs значительная часть будет «отбракована» в ходе испытаний, и на это потребуется много времени. То же касается и RFs, при этом разработки стимуляторов имеют меньше примеров и потребуют значительной креативности.
В этой связи интересна позиция авторов, предлагающих провести испытания противовирусной активности препаратов, применяемых для лечения других заболеваний – как инфекционных, так и неинфекционных. Примеры такого рода имеются в истории разработки антиретровирусных средств; например, легендарный азидотимидин был изобретен как препарат с широким антибактериальным и противопаразитарным эффектом [60]. Есть сведения о том, что препараты для коррекции липидного обмена (статины) оказываются эффективны в предотвращении роста вирусной нагрузки у ВИЧ-инфицированных пациентов [61], хотя механизм этого эффекта остается непонятен. Очевидное достоинство «старых» препаратов заключается в том, что для их внедрения не потребуются долгие годы предварительных клинических испытаний.
Наконец, перспективной представляется мысль о том, чтобы сделать объектом антиретровирусного воздействия не сами вирусные и клеточные белки, а возможность их контакта – так называемый интерактом (interactome) «вирус–хозяин» [7], и обеспечить таким образом прерывание ключевых взаимодействий хозяина с вирусом. Примером такого рода могут быть описанные выше аллостерические ингибиторы LEDGINs, играющие роль «распорки» между вирусным белком IN и клеточным фактором LEDGF.
Итак, участие различных клеточных образований в патогенезе ВИЧ-инфекции становится все более очевидным. В настоящем обзоре сделана попытка кратко представить некоторые из внутриклеточных и молекулярных взаимодействий вирусных и клеточных белков, известных на сегодняшний день. Идентификация и исследование множества белков клетки, эксплуатируемых вирусом на всех этапах жизненного цикла, представляет большие возможности для АРТ. Клеточные мишени особенно привлекательны тем, что практически не подвержены мутационным изменениям, а значит, проблема лекарственной устойчивости для них существенно менее актуальна, чем для вирусных мишеней. Тем не менее, прежде чем имеющиеся знания станут основой для разработки новых лекарственных препаратов, нужно получить ответы на многие вопросы. Следует выяснить, насколько явления и процессы, наблюдаемые пока в основном в условиях in vitro, соответствуют реальности in vivo; какие из активностей клеточных белков могут быть ингибированы без заметного ущерба для метаболизма самой клетки; функцию каких факторов можно «выключить» без ощутимых физиологических последствий. За результатами этих исследований мы будем с интересом наблюдать в ближайшие годы.
Об авторах
Марина Ридовна Бобкова
ФГБНУ «Научно-исследовательский институт вакцин и сывороток им. И.И. Мечникова»
Автор, ответственный за переписку.
Email: mrbobkova@mail.ru
ORCID iD: 0000-0001-5481-8957
д-р биол. наук, главный специалист лаборатории биологии лентивирусов ФГБНУ «НИИ вакцин и сывороток им. И.И. Мечникова», Москва, Россия
Россия, 105064, г. МоскваСписок литературы
- Roa-Linares V.C., Escudero-Florez M., Vicente-Manzanares M., Gallego-Gomez J.C. Host cell targets for unconventional antivirals against RNA viruses. Viruses. 2023; 15(3): 776. https://doi.org/10.3390/v15030776
- Bolinger C., Boris-Lawrie K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology. 2009; 6: 8. https://doi.org/10.1186/1742-4690-6-8
- Colomer-Lluch M., Ruiz A., Moris A., Prado J.G. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front. Immunol. 2018; 9: 2876. https://doi.org/10.3389/fimmu.2018.02876
- Ghimire D., Rai M., Gaur R. Novel host restriction factors implicated in HIV-1 replication. J. Gen. Virol. 2018; 99(4): 435–46. https://doi.org/10.1099/jgv.0.001026
- Puhl A.C., Garzino Demo A., Makarov V.A., Ekins S. New targets for HIV drug discovery. Drug Discov. Today. 2019; 24(5): 1139–47. https://doi.org/10.1016/j.drudis.2019.03.013
- Schaller T., Herold N. The early bird catches the worm – can evolution teach us lessons in fighting HIV? Curr. HIV Res. 2016; 14(3): 183–210. https://doi.org/10.2174/1570162x14999160224094914
- Shukla E., Chauhan R. Host-HIV-1 interactome: a quest for novel therapeutic intervention. Cells. 2019; 8(10): 1155. https://doi.org/10.3390/cells8101155
- Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008; 319(5865): 921–6. https://doi.org/10.1126/science.1152725
- Goffinet C. Cellular antiviral factors that target particle infectivity of HIV-1. Curr. HIV Res. 2016; 14(3): 211–6. https://doi.org/10.2174/1570162x14666151216145521
- Alvarez V., Lopez-Larrea C., Coto E. Mutational analysis of the CCR5 and CXCR4 genes (HIV-1 co-receptors) in resistance to HIV-1 infection and AIDS development among intravenous drug users. Hum. Genet. 1998; 102(4): 483–6. https://doi.org/10.1007/s004390050726
- Cohn S.K. Jr., Weaver L.T. The black death and AIDS: CCR5-Delta32 in genetics and history. QJM. 2006; 99(8): 497–503. https://doi.org/10.1093/qjmed/hcl076
- Dean M., Carrington M., Winkler C., Huttley G.A., Smith M.W., Allikmets R., et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996; 273(5283): 1856–62. https://doi.org/10.1126/science.273.5283.1856
- Tsui C.K., Gupta A., Bassik M.C. Finding host targets for HIV therapy. Nat. Genet. 2017; 49(2): 175–6. https://doi.org/10.1038/ng.3777
- Park R.J., Wang T., Koundakjian D., Hultquist J.F., Lamothe-Molina P., Monel B., et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Gen. 2017; 49(2): 193–203. https://doi.org/10.1038/ng.3741
- Blumenthal R., Durell S., Viard M. HIV entry and envelope glycoprotein-mediated fusion. J. Biol. Chem. 2012; 287(49): 40841–9. https://doi.org/10.1074/jbc.r112.406272
- Hadpech S., Moonmuang S., Chupradit K., Yasamut U., Tayapiwatana C. Updating on roles of HIV intrinsic factors: A review of their antiviral mechanisms and emerging functions. Intervirology. 2022; 65(2): 67–79. https://doi.org/10.1159/000519241
- Gonzalez-Enriquez G.V., Escoto-Delgadillo M., Vazquez-Valls E., Torres-Mendoza B.M. SERINC as a Restriction Factor to Inhibit Viral Infectivity and the Interaction with HIV. J. Immunol. Res. 2017; 2017: 1548905. https://doi.org/10.1155/2017/1548905
- Tedbury P.R., Sarafianos S.G. Exposing HIV’s weaknesses. J. Biol. Chem. 2017; 292(14): 6027–8. https://doi.org/10.1074/jbc.h117.777714
- Lopez Hernandez M., Lopez De Lucio S. Accessory regulatory proteins of HIV-1 and host restriction factors interactions. Biomed. J. Sci. Tech. Res. 2020; 31(4): 24308–12. https://doi.org/10.26717/BJSTR.2020.31.005120
- Huérfano S., Šroller V., Bruštíková K., Horníková L., Forstová J. The interplay between viruses and host DNA sensors. Viruses. 2022; 14(4): 666. https://doi.org/10.3390/v14040666
- Yin X., Langer S., Zhang Z., Herbert K.M., Yoh S., König R., et al. Sensor sensibility-HIV-1 and the innate immune response. Cells. 2020; 9(1): 254. https://doi.org/10.3390/cells9010254
- Yamashita M., Engelman A.N. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017; 25(9): 741–55. https://doi.org/10.1016/j.tim.2017.04.004
- Ramdas P., Sahu A.K., Mishra T., Bhardwaj V., Chande A. From entry to egress: strategic exploitation of the cellular processes by HIV-1. Front. Microbiol. 2020; 11: 559792. https://doi.org/10.3389/fmicb.2020.559792
- Malim M.H., Bieniasz P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012; 2(5): a006940. https://doi.org/10.1101/cshperspect.a006940
- van Manen D., Rits M.A., Beugeling C., van Dort K., Schuitemaker H., Kootstra N.A. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008; 4(2): e18. https://doi.org/10.1371/journal.ppat.0040018
- Kim K., Dauphin A., Komurlu S., McCauley S.M., Yurkovetskiy L., Carbone C., et al. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 2019; 4(12): 2044–51. https://doi.org/10.1038/s41564-019-0592-5
- Harris R.S., Hultquist J.F., Evans D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012; 287(49): 40875–83. https://doi.org/10.1074/jbc.r112.416925
- Engelman A., Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008; 4(3): e1000046. https://doi.org/10.1371/journal.ppat.1000046
- Renzi G., Carta F., Supuran C.T. The integrase: an overview of a key player enzyme in the antiviral scenario. Int. J. Mol. Sci. 2023; 24(15): 12187. https://doi.org/10.3390/ijms241512187
- Lee M.S., Craigie R. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl Acad. Sci. USA. 1998; 95(4): 1528–33. https://doi.org/10.1073/pnas.95.4.1528
- Bin Hamid F., Kim J., Shin C.G. Cellular and viral determinants of retroviral nuclear entry. Can. J. Microbiol. 2016; 62(1): 1–15. https://doi.org/10.1139/cjm-2015-0350
- Tingey M., Li Y., Yu W., Young A., Yang W. Spelling out the roles of individual nucleoporins in nuclear export of mRNA. Nucleus. 2022; 13(1): 170–93. https://doi.org/10.1080/19491034.2022.2076965
- Endsley M.A., Somasunderam A.D., Li G., Oezguen N., Thiviyanathan V., Murray J.L., et al. Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology. 2014; 454-455: 60–6. https://doi.org/10.1016/j.virol.2014.02.006
- Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010; 7(3): 221–33. https://doi.org/10.1016/j.chom.2010.02.007
- Kane M., Yadav S.S., Bitzegeio J., Kutluay S.B., Zang T., Wilson S.J., et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013; 502(7472): 563–6. https://doi.org/10.1038/nature12653
- Wei W., Guo H., Ma M., Markham R., Yu X.F. Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine. 2016; 8: 230–6. https://doi.org/10.1016/j.ebiom.2016.04.020
- Lelek M., Casartelli N., Pellin D., et al. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015; 6: 6483. https://doi.org/10.1038/ncomms7483
- Demeulemeester J., De Rijck J., Gijsbers R., Debyser Z. Retroviral integration: Site matters: Mechanisms and consequences of retroviral integration site selection. Bioessays. 2015; 37(11): 1202–14. https://doi.org/10.1002/bies.201500051
- Bedwell G.J., Engelman A.N. Factors that mold the nuclear landscape of HIV-1 integration. Nucleic Acids Res. 2021; 49(2): 621–35. https://doi.org/10.1093/nar/gkaa1207
- Saito A., Henning M.S., Serrao E., Dubose B.N., Teng S., Huang J., et al. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J. Virol. 2016; 90(15): 6918–35. https://doi.org/10.1128/jvi.00019-16
- Maillot B., Lévy N., Eiler S., Crucifix C., Granger F., Richert L., et al. Structural and functional role of INI1 and LEDGF in the HIV-1 preintegration complex. PloS One. 2013; 8(4): e60734. https://doi.org/10.1371/journal.pone.0060734
- Lapaillerie D., Lelandais B., Mauro E., Lagadec F., Tumiotto C., Miskey C., et al. Modulation of the intrinsic chromatin binding property of HIV-1 integrase by LEDGF/p75. Nucleic Acids Res. 2021; 49(19): 11241–56. https://doi.org/10.1093/nar/gkab886
- Christ F., Voet A., Marchand A., Nicolet S., Desimmie B.A., Marchand D., et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010; 6(6): 442–8. https://doi.org/10.1038/nchembio.370
- Desimmie B.A., Schrijvers R., Demeulemeester J., Borrenberghs D., Weydert C., Thys W., et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology. 2013; 10: 57. https://doi.org/10.1186/1742-4690-10-57
- Le Rouzic E., Bonnard D., Chasset S., Bruneau J.M., Chevreuil F., Le Strat F., et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology. 2013; 10: 144. https://doi.org/10.1186/1742-4690-10-144
- Vranckx L.S., Demeulemeester J., Saleh S., Boll A., Vansant G., Schrijvers R., et al. LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine. 2016; 8: 248–64. https://doi.org/10.1016/j.ebiom.2016.04.039
- Bruggemans A., Vansant G., Balakrishnan M., Mitchell M.L., Cai R., Christ F., et al. GS-9822, a preclinical LEDGIN candidate, displays a block-and-lock phenotype in cell culture. Antimicrob. Agents Chemother. 2023; 65(5): e02328-20. https://doi.org/10.1128/aac.02328-20
- Debyser Z., Bruggemans A., Van Belle S., Janssens J., Christ F. LEDGINs, inhibitors of the interaction between HIV-1 integrase and LEDGF/p75, are potent antivirals with a potential to cure HIV infection. Adv. Exp. Med. Biol. 2021;1322: 97–114. https://doi.org/10.1007/978-981-16-0267-2_4
- Romani B., Allahbakhshi E. Underlying mechanisms of HIV-1 latency. Virus Genes. 2017; 53(3): 329–39. https://doi.org/10.1007/s11262-017-1443-1
- Wang S., Qiu L., Yan X., Jin W., Wang Y., Chen L., et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology. 2012; 55(3): 730–41. https://doi.org/10.1002/hep.24809
- Бобкова М.Р. Латентность ВИЧ. М.: Человек; 2021.
- Кузнецова А.И., Громов К.Б., Киреев Д.Е., Шлыкова А.В., Лопатухин А.Э., Казеннова Е.В. и др. Анализ особенностей белка Tat вируса иммунодефицита человека 1 типа суб-субтипа А6 (Retroviridae: Orthoretrovirinae: Lentivirus: Human immunodefciency virus-1). Вопросы вирусологии. 2021; 66(6): 452–64. https://doi.org/10.36233/0507-4088-83 https://elibrary.ru/cmzgyc (in Russian)
- Nchioua R., Bosso M., Kmiec D., Kirchhoff F. Cellular factors targeting HIV-1 transcription and viral RNA transcripts. Viruses. 2020; 12(5): 495. https://doi.org/10.3390/v12050495
- Mousseau G., Valente S.T. Role of host factors on the regulation of tat-mediated HIV-1 transcription. Curr. Pharm. Des. 2017; 23(28): 4079–90. https://doi.org/10.2174/1381612823666170622104355
- Naji S., Ambrus G., Cimermančič P., Reyes J.R., Johnson J.R., Filbrandt R., et al. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol. Cell. Proteomics. 2012; 11(4): M111.015313. https://doi.org/10.1074/mcp.m111.015313
- Lerner G., Weaver N., Anokhin B., Spearman P. Advances in HIV-1 assembly. Viruses. 2022; 14(3): 478. https://doi.org/10.3390/v14030478
- Rose K.M. When in need of an ESCRT: The nature of virus assembly sites suggests mechanistic parallels between nuclear virus egress and retroviral budding. Viruses. 2021; 13(6): 1138. https://doi.org/10.3390/v13061138
- Sauter D. Counteraction of the multifunctional restriction factor tetherin. Front. Microbiol. 2014; 5: 163. https://doi.org/10.3389/fmicb.2014.00163
- McNatt M.W., Zang T., Bieniasz P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013; 9(4): e1003299. https://doi.org/10.1371/journal.ppat.1003299
- Pattishal K. Discovery and development of Zidovudine as the cornerstone of therapy to control human immunodeficiency virus infection. In: Adams J., Merluzzi V.J., eds. The Search for Antiviral Drugs: Case Histories from Concept to Clinic. Boston, MA: Birkhäuser; 1993.
- Drechsler H., Ayers C., Cutrell J., Maalouf N., Tebas P., Bedimo R. Current use of statins reduces risk of HIV rebound on suppressive HAART. PLoS One. 2017; 12(3): e0172175. https://doi.org/10.1371/journal.pone.0172175
Дополнительные файлы

